
International Internal Medicine Journal(IIMJ)
ISSN: 2837-4835 | DOI: 10.33140/IIMJ
Impact Factor: 1.02

Research Article - (2025) Volume 3, Issue 4
Mixed Connective Tissue Disease: A Rare Disease with Many Faces
Received Date: Jun 16, 2025 / Accepted Date: Jul 28, 2025 / Published Date: Aug 08, 2025
Copyright: ©©2025 Chaimaa Zeroual*, Zeroual, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Citation: Zeroual, C., Aallam, A., Mourabit, S., Moudatir, M., Echchilali, K., et al. (2025). Mixed Connective Tissue Disease: A Rare Disease with Many Faces. Int Internal Med J, 3(4), 01-07.
Abstract
Introduction: Mixed connective tissue disease (MCTD) is a rare autoimmune disease characterized by overlapping clinical manifestations of several systemic connective tissue diseases and the presence of anti-U1RNP antibodies. This study aims to describe the clinical, immunological, and evolutionary features of patients with MCTD.
Methods: A retrospective descriptive study conducted over 5 years (2020–2024) in the internal medicine department of Ibn Rochd University Hospital Center in Casablanca, including 9 female patients meeting Kasukawa’s criteria.
Results: All patients were female, with a mean age at diagnosis of 41.3 years. Raynaud’s phenomenon was the most frequent inaugural sign. Pulmonary involvement was present in 66.6% of cases, with one-third of these patients exhibiting fibrotic changes. Anti-U1RNP antibodies were positive in 100% of patients. Evolution towards a differentiated connective tissue disease (lupus or systemic sclerosis) was observed in 77.7% of cases. The overall prognosis was favorable, despite one death related to a paraneoplastic syndrome.
Conclusion: MCTD remains a rare disease with variable clinical expression, requiring particular diagnostic vigilance. Early identification of evolving forms and multidisciplinary monitoring are essential to prevent complications and guide treatment
Keywords
Mixed Connective Tissue Disease, Sharp's Syndrome, Anti-U1RNP, Ibn Rochd University Hospital Center, Casablanca
Introduction
Mixed connective tissue disease (MCTD) is a rare systemic autoimmune disorder characterized by an overlap of several connective tissue diseases, notably systemic lupus erythematosus and systemic sclerosis, in the presence of high-titer anti-U1- RNP antibodies [1-3]. Its clinical presentation is heterogeneous, frequently involving articular, muscular, cutaneous, pulmonary, or digestive manifestations, which often complicates and delays diagnosis [4]. Despite advances in the field, MCTD remains a clinical entity with still ill-defined boundaries, necessitating deeper insight to enhance patient management. The objective of this study is to report the experience of the Internal Medicine Department at Ibn Rochd University Hospital Center in Casablanca regarding this pathology over a five-year period.
Methods
This retrospective descriptive study was conducted from January 2020 to December 2024. The analysis included 9 female patients diagnosed with MCTD according to Kasukawa’s criteria. Data were collected through a thorough review of the patients’ medical records encompassing hospital admissions.
Results
Demographic Data
Among the 1921 patients followed in internal medicine during the study period, 9 cases of MCTD were identified, representing a prevalence of 0.4%. All patients were female, with a mean age at diagnosis of 41.3 years.
Clinical Presentation
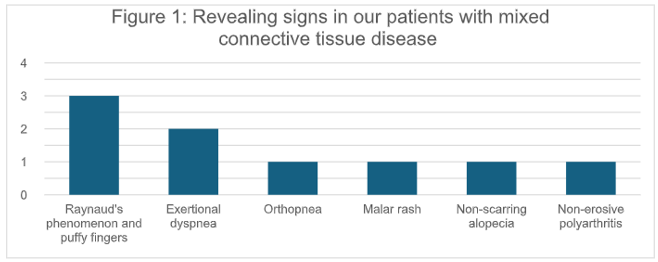
The most frequent inaugural presentation was Raynaud's phenomenon accompanied by puffy fingers (33.3%). Other notable clinical signs observed included (Figure 1):
- Exertional dyspnea (Grade 3 on the modified Medical Research Council (mMRC) scale): 2 patients
- Orthopnea with abundant pericardial effusion: 1 patient - Malar rash (erythema in vespertilio): 1 patient
- Non-scarring alopecia: 1 patient
- Non-erosive polyarthritis: 1 patient
Associated Autoimmune Diseases
The presence of other autoimmune comorbidities was significant (Figure 2):
- Hashimoto's thyroiditis: 6 patients (66,6%)
- Sjögren's disease: 2 patients (22.2%)
- Immune thrombocytopenia: 1 patient (11,1%)
- Antiphospholipid syndrome (APS): 1 patient (11,1%)

Figure 2 : Autoimmune Diseases Associated to Mixed Connective Tissue Diseases in Our Series
Ancillary Investigations
In capillaroscopy, all patients exhibited distal microangiopathy, with severe findings (e.g., capillary deserts, hemorrhages) in 33.3% of cases (Figure 3).

Immunologically, antinuclear antibodies (ANA) were positive in 100% of patients (titer ≥ 1/640). The immunofluorescence pattern was speckled in 66.6% and a mixed homogeneous and speckled pattern in 33.3% (Figure 4). Anti-U1RNP antibodies were positive in 100% of patients, with strong positivity noted in 33.3% (Figure 5).

Organ Involvement
8 patients presented with pulmonary hypertension (PHT) in echocardiogram (Figure 6). Whereas confirmed PHT was present in only 2 patients. Thoracic computed tomography scan revealed no involvement in 33.3% of cases, non-fibrosing interstitial lung disease in 33.3%, and pulmonary fibrosis in 33.3% (Figure 7). Pulmonary function tests indicated restrictive ventilatory defects in 22.2% and mixed ventilatory defects in 22.2%.

Therapeutic Management
All patients received a calcium channel blocker. In addition, the following treatments were administered:
-Corticosteroids and mycophenolate mofetil: 55.5%
-Hydroxychloroquine: 44.4%
Evolution and Prognosis
The disease course demonstrated varied differentiation patterns (Figure 8):
- Differentiation towards systemic sclerosis: 33.3% (including one paraneoplastic case)
- Differentiation towards systemic lupus erythematosus: 44.4%
- Not individualized MCTD: 22.2%
The overall outcome was favorable in 88.8% of cases. Unfortunately, one patient (11.1%) succumbed to the disease, specifically due to a paraneoplastic form of systemic sclerosis.

Discussion
MCTD, also called Sharp’s syndrome, is an autoimmune entity defined by the coexistence of symptoms belonging to several systemic connective tissue diseases (systemic lupus erythematosus, systemic sclerosis, polymyositis) and by the presence of anti- U1RNP antibodies. Diagnosis is based on criteria sets such as those by Sharp, Kasukawa, Alarcón-Segovia, Kahn and Tanaka (Table 1) [1,5- 8].
|
Criteria |
Key Criteria |
Clinical Features |
Immunological Features |
Diagnostic Requirements |
Sensitivity / Specificity |
|
Sharp Criteria [1] |
Major criteria: severe myositis, pulmonary involvement (DLCO <70%, PAH, vascular lesions), Raynaud's phenomenon (RP) or esophageal hypomotility, edema of hands/sclerodactyly, anti-ENA antibodies ≥1:10,000 with anti-nRNP positive and anti-Sm negative |
Myositis, RP, swollen hands, pulmonary involvement |
High titer anti-U1-RNP antibody, anti- ENA positive, anti-Sm negative |
4 major criteria + anti- RNP titer >1:4000; or 2 major + 2 minor + anti- nRNP ≥1:1000 |
Sensitivity 57.7%, Specificity 90% |
|
Kahn Criteria [7] |
Serological: Anti-nRNP >1:1600 |
RP, synovitis, myositis, swollen fingers |
Anti-nRNP antibody titer >1:1600 |
Serological criterion + RP + ≥2 clinical criteria |
Sensitivity 52.3%, Specificity 99.4% |
|
Alarcón- Segovia Criteria [6] |
Serological: Anti-nRNP >1:1600 |
Edema of hands, synovitis, myositis, RP, acrosclerosis |
Anti-nRNP antibody titer >1:1600 |
Serological + ≥3 clinical criteria (including synovitis or myositis) |
Sensitivity 69.4%, Specificity 99.4% |
|
Kasukawa Criteria [5] |
Common symptoms: RP, swollen fingers/hands, anti-nRNP positive |
SLE-like (polyarthritis, rash, lymphadenopathy, pericarditis); SSc-like (sclerodactyly, ILD, esophageal dysmotility); PM-like (muscle weakness, elevated CPK, myogenic EMG) |
Anti-nRNP positive |
≥1 common symptom + anti-nRNP + ≥1 mixed symptom from at least two categories (SLE-like, SSc-like, PM-like) |
Sensitivity 77.5%, Specificity 92.2% |
|
Tanaka Criteria [8] |
I. Common: RP, puffy fingers/swollen hands; II. Immunological: Anti- U1RNP positive; III. Characteristic organ: PAH, aseptic meningitis, trigeminal neuropathy; IV. Overlapping manifestations (SLE- like, SSc-like, PM/DM-like features) |
Includes systemic features of SLE, systemic sclerosis, polymyositis |
Anti-U1RNP positivity required |
At least one common + immunological + one characteristic organ involvement OR one from overlapping manifestations in 2+ categories |
Sensitivity 90.6%, Specificity 98.4% |
Table 1 : The different diagnostic criteria for MCTD based on established classifications from the literature. Abbreviations: RP: Raynaud’s Phenomenon ; Anti-nRNP: Anti-U1 ribonucleoprotein antibodies ; PAH: Pulmonary arterial hypertension ; SLE: Systemic lupus erythematosus ; SSc: Systemic sclerosis ; PM/DM: Polymyositis/Dermatomyositis ; DLCO: Diffusing capacity for carbon monoxide ; CPK: Creatine phosphokinase.
Although recognized since the 1970s , MCTD remains uncommon, representing less than 2% of systemic connective tissue diseases in most series . In our study, it accounted for 0.4% of patients followed in internal medicine, confirming its rarity. The female predominance is well-established: in the large cohort by Burdt et al. (n = 91), 92% of patients were women, as was the case in the Norwegian study by Gunnarsson et al. (n = 147), with 89% female patients [9,10]. Our series is consistent, with 100% female patients. The mean age at diagnosis generally ranges between 30 and 50 years [9,10]. In our series, the mean age at diagnosis was 41.3 years, falling within this range.
Raynaud’s phenomenon is often the inaugural sign, reported in 80 to 100% of cases. In our study, it was present in one-third of patients at onset. It is frequently associated with capillaroscopic abnormalities, as we also observed in all of our patients. Severe forms (microangiopathy, capillary dropout) were present in 33.3%, comparable to what Cutolo et al. report in systemic connective tissue diseases with early vascular involvement [11].
Arthralgias and non-erosive polyarthritis occur in 50 to 80% of cases, often rheumatoid-like but without joint destruction [12]. In our cohort, only one patient (11%) presented with arthropathy. Malar rash (11%) and non-scarring alopecia (11%) are more characteristic manifestations of lupus, also found in our series.
The coexistence of autoimmune diseases is frequent. Autoimmune thyroiditis is found in about 30 to 40% of MCTD cases [13,14]. In our study, this proportion was even higher (66.6%), which might reflect a specificity of our population or a recruitment bias. Sjögren’s syndrome is also reported in MCTD, found in 22.2% of our patients [15].
Antinuclear antibodies (ANA) positivity is constant in MCTD, generally at high titers (≥1/640), with a speckled or mixed pattern [16]. Our data confirm this tendency: all cases were positive, with 77.7% having a titer of 1/1280. The anti-U1RNP antibody is the central immunological marker [17]. It was strongly positive in all our patients, in line with Kasukawa’s criteria. Other autoantibodies (anti-dsDNA, anti-Ro/SSA, anti-Scl70) may be transiently present but are not specific (Table 2) [18,19].
|
Autoantibody |
Clinical/Diagnostic Notes |
|
Anti-U1-RNP |
Hallmark antibody; key diagnostic marker for MCTD |
|
Antinuclear Antibody (ANA) |
Positive ANA with speckled pattern common |
|
Anti-Sm |
Can develop over time; associated with overlapping autoimmune features |
|
Rheumatoid Factor (RF) |
Associated with arthritis features and sometimes with erosive arthritis |
|
Anti-dsDNA |
More typical of systemic lupus but seen in MCTD overlap patients |
|
Anti-Ro/SSA |
Seen variably; may indicate overlap with Sjögren’s or lupus features |
|
Anti-CCP |
Linked to erosive arthritis in a subset of MCTD patients |
|
Anti-PM-Scl |
Suggests overlap with myositis or scleroderma components |
|
Anti-Ku |
Associated with overlap syndromes involving myositis and lung disease |
Table 2: Clinical/Diagnostic Notes of Autoantibodies in MCTD
Pulmonary involvement is the main cause of death in MCTD. Pulmonary fibrosis (PF) is found in 20 to 40% of cases depending on the series, with a strong association to scleroderma-like progression [20]. In our study, one-third of patients already had PF at diagnosis, demonstrating the potential severity of respiratory involvement. Restrictive or mixed ventilatory defects, found in 44% of our patients, are also well described in pulmonary function tests of patients with MCTD.
Pulmonary arterial hypertension (PAH) affects 10 to 20% of patients in various series (up to 40% in advanced forms). PAH is the most serious life-threatening complication, making regular screening for this condition crucial [21]. Its detection via echocardiography and confirmation by right heart catheterization is essential [22]. In our cohort, 22.2% of patients had confirmed PAH.
Prevalence of cardiac involvement varied from 13% to 65% in MCTD cases, and pericarditis is the most common cardiac diagnosis with a prevalence of 30% and 43% [23]. One of our patients presented with orthopnea and a large pericardial effusion, which is consistent. Neurological and renal involvements are rarer and were not found in our series.
A major feature of MCTD is its nosological instability. Several longitudinal studies show that about 30 to 70% of patients evolve over time toward a differentiated connective tissue disease, most often systemic lupus or systemic sclerosis [24,25]. In our series, 77.7% of patients had already evolved: 44.4% toward lupus, 33.3% toward systemic sclerosis (including one paraneoplastic case), and 22.2% remained undifferentiated. This progression suggests that MCTD could represent an intermediate or early state of other connective tissue diseases rather than a stable entity. This hypothesis is reinforced by the clinical, immunological, and therapeutic evolution observed over time.
Treatment for MCTD depends on the type of manifestations. Corticosteroids are widely used to control inflammatory phases, often combined with immunosuppressants (azathioprine, mycophenolate mofetil, methotrexate) depending on organ involvement [26]. In our study, 55.5% received corticosteroids and mycophenolate. Hydroxychloroquine is recommended for mild articular or cutaneous involvement [27].
For interstitial lung disease, particularly early or moderate forms, several studies have demonstrated the efficacy of mycophenolate mofetil and cyclophosphamide in stabilizing respiratory function and even improving functional parameters [28].
Conclusion
MCTD is a rare and complex autoimmune disorder that poses significant diagnostic challenges due to the absence of highly specific criteria. Enhancing the standardization of immunological and clinical assessment tools is essential to enable earlier detection. Physician’s main goal remains improving patient monitoring and better anticipating potential complications. Looking ahead, ongoing research hold promise for more precise diagnostic methods and targeted therapies, offering hope for management of MCTD in the future.
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All procedures performed in this study are in accordance with the ethical standards of the institutional and/or national research comittee(s). Written informed consent was obtained from the patients for publication of this case series.
References
- Sharp, G. C., Irvin, W. S., Tan, E. M., Gould, R. G., & Holman, H. R. (1972). Mixed connective tissue disease-an apparently distinct rheumatic disease syndrome associated with a specific antibody to an extractable nuclear antigen (ENA). The American journal of medicine, 52(2), 148-159.
- Gunnarsson, R., Hetlevik, S. O., Lilleby, V., & Molberg, Ø. (2016). Mixed connective tissue disease. Best practice & research Clinical rheumatology, 30(1), 95-111.
- Mahler M, Kessenbrock K, Szmyrka M, et al. Diagnostic utility of antibodies to U1-RNP in suspected connective tissue disease: a systematic review. Autoimmun Rev. 2015;14(3):268–273.
- Charoenlap C, et al. (2020). Clinical and immunological profile of mixed connective tissue disease: Comparison of Sharp, Kasukawa, Alarcón-Segovia, and Kahn criteria. Clin Rheumatol. ;39(3):781-787.
- KASUKAWA, R. (1987). Diagnosis of mixed connective tissue disease: preliminary diagnostic criteria for classification of mixed connective tissue disease. Mixed connective tissue disease and anti-nuclear antibodies, 41-48.
- Alarcon-Segovia, D. (1987). Classification and diagnostic criteria for mixed connective tissue disease. Mixed connective tissue disease and anti-nuclear antibodies.
- Kahn MF. Mixed connective tissue disease. In: Decker JL, Klippel JH, editors. Rheumatology. 1983;21(4):178–182.
- Tanaka, Y., Kuwana, M., Fujii, T., Kameda, H., Muro, Y., Fujio, K., ... & Mori, M. (2021). 2019 Diagnostic criteria for mixed connective tissue disease (MCTD): from the Japan research committee of the ministry of health, labor, and welfare for systemic autoimmune diseases. Modern rheumatology, 31(1), 29-33.
- Burdt MA, Hoffman RW, Deutscher SL, Wang GS, Johnson JC, Sharp GC. (1999). Longitudinal clinical study of patients with mixed connective tissue disease. Arthritis Rheum.;42(5):899-909.
- Gunnarsson R, Molberg Ø, Gilboe IM, Gran JT. (2011). The prevalence and incidence of mixed connective tissue disease: a national multicentre survey of Norwegian patients. Ann Rheum Dis;70(6):1047–1051.
- Cutolo M, Smith V, Sulli A, et al. (2023). Microvascular damage in autoimmune connective tissue diseases. Clin Exp Rheumatol;41(5):836-842.
- Bennett, R. M., & O'Connell, D. J. (1978). The arthritis of mixed connective tissue disease. Annals of the Rheumatic Diseases, 37(5), 397-403.
- Salah, R. B., Kacem, F. H., Soomauro, S., Chouaib, S., Frikha, F., Charfi, N., ... & Bahloul, Z. (2022). Autoimmune thyroiditis associated with autoimmune diseases. Electronic Journal of General Medicine, 19(6).
- Ban Y, Tozaki T, Taniyama M, Takasu N, Ban Y. (1995). Prevalence of autoimmune thyroid disease in patients with mixed connective tissue disease. J Rheumatol; 22(5):1013-6.
- Usuba, F. S., Lopes, J. B., Fuller, R., Yamamoto, J. H., Alves,M. R., Pasoto, S. G., & Caleiro, M. T. C. (2014). Sjögren's syndrome: an underdiagnosed condition in mixed connectivetissue disease. Clinics, 69(3), 158-162.
- Pahor, A., Krajnc, I., Gorenjak, M., & Holc, I. (1998). The clinical significance of antinuclear antibodies in connective tissue disease. Wiener Klinische Wochenschrift, 110(9), 338- 341.
- Elhani, I., Khoy, K., Mariotte, D., Comby, E., Marcelli, C., Le Mauff, B., ... & de Boysson, H. (2023). The diagnostic challenge of patients with anti-U1-RNP antibodies. Rheumatology international, 43(3), 509-521.
- Abdelgalil Ali Ahmed, S., Adam Essa, M. E., Ahmed, A. F., Elagib, E. M., Ahmed Eltahir, N. I., Awadallah, H., ... & Ebad, M. A. B. (2021). Incidence and clinical pattern of mixed connective tissue disease in Sudanese patients at Omdurman military hospital: hospital-based study. Open Access Rheumatology: Research and Reviews, 333-341.
- Maikap, D., Deosale, S., Singh, P., Panda, S. S., & Padhan,P. (2023). A comparative study of mixed connective tissue disease and overlap syndromes–A single-center study from India. Indian Journal of Rheumatology, 18(3), 192-198.
- Gono T, Kawaguchi Y, Satoh T, Kuwana M, Katsumata Y, Takagi K, et al. Clinical manifestations of pulmonary fibrosis in patients with mixed connective tissue disease. Rheumatology (Oxford). 2010;49(5):895–900.
- Aringer, M., & Smolen, J. S. (2007). Mixed connective tissue disease: what is behind the curtain?. Best Practice & Research Clinical Rheumatology, 21(6), 1037-1049.
- Zanatta, E., Polito, P., Famoso, G., Larosa, M., De Zorzi,E., Scarpieri, E., ... & Doria, A. (2019). Pulmonary arterial hypertension in connective tissue disorders: Pathophysiology and treatment. Experimental biology and medicine, 244(2), 120-131.
- Ungprasert, P., Wannarong, T., Panichsillapakit, T., Cheungpasitporn, W., Thongprayoon, C., Ahmed, S., & Raddatz, D. A. (2014). Cardiac involvement in mixed connective tissue disease: a systematic review. International journal of cardiology, 171(3), 326-330.
- Zahn S, Barchet W, Rehkämper C, Huber M, Thiel A, Schmidgen T, et al. (2011). Predictors of disease progression in mixed connective tissue disease. Clin Exp Rheumatol;29(3):499–504.
- Ungprasert P, Wannarong T, Panichsillapakit T, Cheungpasitporn W, Spanuchart I, Suksaranjit P. (2014). Clinical features and outcomes of mixed connective tissue disease: a comparative study with systemic lupus erythematosus and systemic sclerosis. Int J Rheum Dis;17(8):834–40.
- Kim, P., & Grossman, J. M. (2005). Treatment of mixed connective tissue disease. Rheumatic Disease Clinics, 31(3), 549-565.
- Izmirly PM, Buyon JP. (2010). Hydroxychloroquine. Rheum Dis Clin North Am;36(1):45–62.
- Tendean, M., Nuriawan, S. A., & Nugroho, P. (2017). Interstitial lung disease in mixed connective tissue disease. Indonesian Journal of Rheumatology, 9(1).