
Journal of Clinical, Medical, and Diagnostic Research(JCMDR)
ISSN: 3065-9817 | DOI: 10.33140/JCMDR


Review Article - (2025) Volume 3, Issue 1
The Role of Polysaccharide Polymers in the Regulation of Angiogenesis and Atherogenic Inflammation in the Wall of Arterial Vessels. Analytical Review
2Professor V.F. Voino-Yasenetsky Krasnoyarsk State Medical University, Russian Federation
3Professor V.F. Voino-Yasenetsky Krasnoyarsk State Medical University, Department of Pathological Ana, Russian Federation
4Professor V.F. Voino-Yasenetsky Krasnoyarsk State Medical University, Head of the Department of Fore, Russian Federation
5Head of the Department of Fundamental Medical Disciplines, Institute of Medicine and Biology, Facult, Russian Federation
Received Date: May 28, 2025 / Accepted Date: Jun 30, 2025 / Published Date: Jul 09, 2025
Copyright: ©Â©2025 Bolshakov Igor Nicolaevich, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Citation: Bolshakov, I. N., Shindyakin, D. V., Bahshyan, V. A., Kirichenko, A. K., Alyabyev, F. V., et al. (2025). The Role of Polysaccharide Polymers in the Regulation of Angiogenesis and Atherogenic Inflammation in the Wall of Arterial Vessels. Analytical Review. J of Cli Med Dia Research, 3(1), 01-69.
Abstract
Damage to peripheral arteries by the atherosclerotic process accounts for about 3% of the world's population, which puts this problem in the category of major social problems. Modified lipoproteins and cholesterol crystals are known to accumulate in the arterial intima and cause the formation of proatherogenic macrophages, foam cells, and inflammation. Defective efferocytosis of apoptotic foam cells leads to the formation of a necrotic core, a sign of failure to resolve inflammation. Resolution of inflammation is mediated by specialized pro-resorption lipid mediators, proteins and signaling gases. Improving the balance between pro- inflammatory and anti-inflammatory factors contributes to the reverse development of local atherogenesis. Active direct intervention in the reconstruction of the adventitial layer of the arterial wall using cholesterol-affinity polysaccharide hydrogels containing a cocktail of growth factors creates conditions for the formation of additional extracellular matrix and reversal of cholesterol mass from the subintimal zone. Creating concentration and electrostatic gradients in the adventitia zone using a hydrogel may be one of the effective ways to degrade early soft atherogenic plaques and locally restore the damaged structure of the vessel wall. Growing scientific interest in the previously insufficiently studied adventitia indicates its important role in angionesis and atherogenesis.
Keywords
Vascular Wall, Angiogenesis, Growth Factors, Atherogenesis, Polysaccharide Polymers, Adventitia
Introduction
Confirming or creating new ideas about the mechanisms of peripheral arterial disease (PAD), mainly of the lower limbs, is a multifaceted and challenging mission for researchers. Peripheral arterial disease affects approximately 3% of the world's population, making it a major social problem [1]. The increase in mortality rate of the population up to 65% due to late diagnosis of the development of critical lower limb ischemia (CLI), despite the high amputations performed, is aggravated by the increased risk of mortality associated with coronary and cerebrospinal diseases [2-4]. The outcome of the final stage of PAD of the lower limbs is 25% mortality and 30% amputations, after which the mortality rate increases significantly [5].
Despite the progress in using various therapeutic strategies for stimulating angiogenesis based on genes, cells, proteins and small molecule drugs in experimental practice for the treatment of PAD, the results of clinical use of these technologies differ significantly from expectations, become unstable and short-term, not to mention low availability for a patient due to the high cost of the drug. Currently, a search is underway for a universal technology for regulating angiogenesis aimed not only at inducing the simultaneous expression of several angiogenic growth factors with the impact of upstream regulators of angiogenesis, but also ensuring effective delivery of regulators to the problematic ischemic zone without losing their activity. This major mission also includes the delivery of viable cell mass in the form of a pool of stem or progenitor cells. Modern ideas about the effectiveness of protection using a system for delivering low-molecular substances, cells or protein growth factors to the lesion zone indicate the prospect of incorporating target products into polymeric biomaterials. Special attention should be paid to modified polysaccharide nano- constructions due to their low toxicity, controlled biodegradability, high biocompatibility, protection of easily hydrolyzed protein molecules in an aggressive environment. In addition to using an effective delivery system, it is necessary to change the technology of local physical (surgical) delivery of substances over a large length of affected main vessels. If the cause of chronic lower limb ischemia is the presence of active atherogenic inflammation and obstruction of blood perfusion by atherogenic plaques, an obvious prospect is to develop a transfer system with high affinity properties for cholesterol and low-density lipoproteins, creating, on the one hand, high accessibility to cells of all three layers of the artery for regulating angiogenesis, and on the other hand, ensuring reverse translation of cholesterol from viable and destroyed cells in the vessel wall.
Mechanisms of Microvessels Formation. Angiogenic Growth Factors
Despite advances in surgical vascular reconstruction in a number of inflammatory and degenerative diseases, in the last few years, attempts to artificial reproduction of the whole vascular hierarchy from large (~6 mm) to small (<0.1 mm) vessels have become a key challenge in the field of tissue engineering research [6]. The field of tissue engineering has evolved in understanding biological mechanisms and engineering strategies to create complex tissues [7]. The challenge of creating scaffolds filled with an integrated functional vascular network remains unresolved.
If positive results were obtained for the creation of thin tissues, then for solid tissues of large mass and high level, technologies were required that could be artificially created and provide blood perfusion, even under conditions of ischemic syndrome. It is not surprising that researchers primarily received two interdependent areas of scientific findings in this scheme: factors of cellular growth activity and biomaterials of natural and synthetic origin. The discovery of angiogenic growth factors has become of crucial importance both for understanding the principles of angiogenesis and for creating vascular networks during regeneration processes, to support cellular development and maintain tissue stability in regenerating areas. It has been established that the presence of several angiogenic growth factors in the affected area increases the number and diameter of newly formed vessels. Modern systems of simultaneous development of several growth factors are usually more effective in inducing angiogenesis compared to sequences with a single release of GF [8-10]. At the same time, the presence of only one growth factor ensures the activity of a number of other growth factors [11,12]. In the case where only one growth factor, for example, VEGF, is used in the model system, the capillary structure is airy [13].
A large number of direct stimulators of neoangiogenesis have been identified: vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF), hepatocyte growth factor (HGF), angiogenin, angiopoietins, platelet-derived growth factor-BB (PDGF-BB) [14]. The group of non-specific stimulators of vascular growth includes insulin-like growth factor 1 (IGF-1), transforming growth factor, tumor necrosis factor, nitric oxide, a number of chemokines, and matrix metalloproteinases (MMPs). These factors induce proliferation of vascular endothelium, its translation into the intimal space with new formation of microvessels. Overexpression of angiogenesis molecules enhances endothelial recruitment and new formation of blood vessels. Migration of closed cells was increased by induction of MMP-2 and MMP -9 [15].
During rest, EC proliferation is inhibited because EC are closely intertwined with vascular endothelial cadherins and supported by pericytes. The onset of the early stage of angiogenesis is stimulated by intermittent processes of mild tissue ischemia. Circulating platelets begin to disrupt the density of interendothelial contacts and adhere to activated endothelial cells, provoking an inflammatory process. As a consequence of such alteration, leukocyte extravasation, changes in vascular permeability and tone, microthrombosis, degradation of the extracellular matrix and angiogenesis occur [16]. As a result of the compensatory response to ischemia and proliferation of endothelial cells, their increased migration and formation of lateral branches from the main vessel occur [17]. The lateral branching of new vessels from the parent vessel is manifested by the formation of apical cells expressing CD31 and CD34 markers and filopodia [18]. This process is accompanied by the formation of endothelial bridges inside the lumen of the main vessel with the inclusion of polarized endothelial cells and the adaptation of their cytoskeleton on both sides to each other, the division of the lumen of the main vessel into subunits. Thus, the microvascular network is laid down [19]. During the recruitment process, the endothelium is divided into two functionally different phenotypes: apical and stem cells. VEGF expression transforms apical cells, which translate into the ECM and peel off thin filopodia from the basement membrane. An increase in VEGF expression by endothelial cells forms a concentration gradient. Neighboring endothelial cells acquire a stem cell phenotype in response to the activation of apical cells. The proliferation of stem cells builds the trunk of a new vessel. Formation of the vessel lumen and initiation of blood perfusion in the functional vascular network occurs with multiple contacts and fusion of filopodia of adjacent cells with the participation of macrophages [20,21]. Formation of endothelial bridges inside the lumen of the main vessel is combined with the mechanism of invagination of endothelial cells into the lumen of the primary vessel (splitting angiogenesis), which leads to expansion of the new vascular network [22]. Thus, translation of active vascular endothelium occurs beyond the vascular wall into the tissue compartment [23-25].
Molecular Structures Influencing ECM Activity
Angiopoetin-1 And Angiopoetin-2 (ANG1 and ANG2)
The destruction of pre-existing blood vessels, proliferation and migration of new cells to form new immature vessels requires the expression of VEGF, FGF-2, HGF, and angiopoietin-2 (ANG- 2) [26]. Stabilization of newly formed vessels, regulation of cell growth and division is achieved by the production of angiopoietin-1 (ANG 1) by ECM cells, followed by triggering the expression of molecules of a group of nonspecific stimulators of angiogenesis [27-30].
Angiopoietins are ligands of the endothelium-specific tyrosine kinase receptor Tie2. Angiopoietin-1 is expressed by mural cells and activates Tie2: it facilitates further recruitment and association of pericytes with newly formed vascular structures, thus creating conditions for the survival of endothelial cells and suppressing VEGF-induced plasma leakage from blood vessels [31,32].
Angiopoietin 2 (ANG 2) is synthesized and stored predominantly in endothelial cells, actively expresses in the ECM when VEGF is released into the environment, and competes with angiopoietin 1 (ANG 1) for binding to the same receptor in the presence of VEGF. Angiopoietin-2 determines endothelial cells to proliferation signals, remodels basement membranes, causes dissociation of pericytes and endothelial cells, and stimulates the migration of endothelial cells [33,34]. This migration accelerates the formation of lateral branches of blood vessels. Direct contact of endothelial cells with pericytes through physical interactions including connexin 43 gap junctions is important for intercellular communication and the participation of adhesion molecules (N-cadherin) [35,36]. Thus, the weakening of contacts between cells occurs with the active participation of ANG 2, which stimulates the extravasation of plasma proteins with the formation of a temporary ECM framework, the induction of pericytes to proteolysis of the ECM with the help of MMPs [37].
Vascular Endothelial Growth Factor (VEGF)
The mammalian VEGF family includes five major ligands (VEGF-A, -B, -C and -D and placental growth factor, PlGF) and three receptors (VEGF-R1, -R2 and -R3). Although the primary role of VEGF-C and -D is to stimulate lymphatic angiogenesis through VEGF-R3, blood vessel growth is primarily coordinated by VEGF-A and -B and PlGF signaling through R1 and R2 [38].
The binding of VEGF to the cell surface occurs through tyrosine kinase receptors (VEGFR) with their trans-phosphorylation, which leads to the launch of signaling pathways for the proliferation and migration of induced cells [39].Vascular endothelial growth factor (VEGF) is an angiogenic glycoprotein that increases vascular permeability, is a selective mitogen for endothelial cells, is synthesized by many polymorphic cells of the blood and interstitial tissue and realizes its neoangiogenic effect independently and together with fibroblast growth factor [40,41].
VEGF family, consisting of 6 isoforms, is responsible for mature and embryonic angiogenesis, lymphangiogenesis, isoforms are expressed in any tissue, of which VEGF-A stimulates the migration and proliferation of endothelial cells in mature angiogenesis [42- 44]. In this case, interendothelial leaks and migration of vascular endothelium are enhanced by blocking the function of vascular cell adhesion protein (VCAM-1) with VEGF.
Activated macrophages, fibroblasts, smooth muscle cells, and mast cells express VEGF in response to the intercellular flow of proinflammatory agents. Endothelial cells have receptors for VEGF, and they actively produce VEGF themselves in a state of hypoxia or anoxia. The production of VEGF by cells of the vascular wall, for example, cardiac muscle, primarily the ECS, and white blood cells penetrating the vascular endothelium stimulate the production of reactive oxygen species (ROS) by the endoplasmic reticulum (ER) cells, which induces the process of autophagy with the simultaneous initiation of the angiogenesis chain [45]. The high angiogenic effect of VEGF-A can be reduced by the inclusion of alternative ligands of the VEGF family in the angiogenesis process. Mild activation profiles are formed due to the competitive participation of VEGF - B in the cell, which indirectly changes the activity of VEGF-R2 and blocks the activation of VEGF-A. Thus, the excess intracellular induction signal is canceled [46]. It has been observed that vessel formation induced by lower levels of VEGF stabilizes more quickly and induces endothelial quiescence [47]. However, acute VEGF deficiency leads to regression of newly formed capillary tubes [48].
A similar effect, regulating the strength of the intracellular signal, can be determined by the native form of VEGF-D, which is capable of switching lymphogenesis to angiogenesis by changing the affinity for VEGF-R2. This ability may be associated with the absence of a heparin-binding domain in the VEGF - D molecule [49].
Placental Growth Factor-2 (PlGF)
Placental growth factor is a member of the VEGF family and is involved in angiogenesis together with VEGF-A and VEGF-B in the activation of VEGFR-1 and VEGFR-2 receptors. When co- expressed in the same cell, VEGF-A and PlGF are able to form a heterodimer. The PlGF2 isoform is capable of exclusively binding to heparin receptors. The formation of the VEGF-A/PlGF2 heterodimer does not reduce vascular density compared to the control. The factor indirectly binds to a wide range of ECM proteins. It is known that the administration of placental growth factor did not cause adverse hemodynamic or systemic inflammatory effects and significantly increased the density of capillaries and arterioles in the ischemic myocardium [50]. However, targeted periadventitial transfer of adenoviruses encoding PlGF2 to the intima of the carotid artery of a hypercholesterolemic rabbit and a subsequent study of the effects of PlGF showed a significant increase in intimal thickness, accumulation of macrophages, and expression of endothelial vascular cell adhesion molecule-1. In addition, adventitial neovascularization was detected in the arteries, a significant increase in the ratio of the intima to the middle vascular layer compared with the vessels of rabbits receiving a normal diet. Thus, local delivery of adenoviral PlGF2 promotes the formation of atherogenic neointima in hypercholesterolemia [51].
Human Telomerase Reverse Transcriptase (hTERT) Human telomerase reverse transcriptase (hTERT) is currently being evaluated in clinical trials for its effectiveness in ischemic vascular disease. hTERT is an important component of telomere elongation. Previous studies have shown that hTERT is one of the downstream effectors of VEGF-A and plays a secondary role in the angiogenic process. Preclinical studies have shown that overexpression of hTERT can enhance the regenerative properties of endothelial progenitor cells (EPCs) and capillary formation in an HLI rat model [52].
Fibroblast Growth Factor (FGF)
There are 22 members of the FGF family with molecular weights ranging from 17 to 34 kDa and shared amino acid identity. FGF is a heparin-binding growth factor, which is expressed in all tissues and acts as a ligand for the FGF receptor (FGF-R). FGFs have various biological functions like in vivo and in in vitro, including roles in angiogenesis. When analyzing the function of basic fibroblast growth factor (bFGF), researchers point to a powerful angiogenic effect emanating from endothelial cell precursors and mature endothelial cells, mesodermal and neuroectodermal cells [53]. bFGF has been shown to promote microvascular formation and is released from the extracellular matrix via heparin sulfate- mediated enzymatic degradation. Under hypoxic conditions, bFGF production is regulated by the remodeling effect of MMP on the perivascular zone of the ECM [54]. bFGF is spontaneously induced after tissue damage and regulates the proliferation, differentiation and migration of endothelial cells [42,55,56].
It is known that the level of endogenous growth factor is insufficient to trigger the process of angiogenesis. For this purpose, researchers add recombinant exogenous bFGF to the delivery system [57]. In addition, in a model of hind limb ischemia in rabbits, the introduction of the FGF -2 plasmid enhanced the growth of capillaries and the formation of collateral blood flow [58]. In a clinical study, intramuscular injection of a plasmid encoding human FGF-1, namely NV1FGF, into patients with CLI reduced the risk of low and high amputations [59].
However, enzymatic protein hydrolysis and rapid destruction of free short-lived growth factor in vivo lead researchers to develop controlled sustained release bFGF delivery systems by incorporating bFGF into tissue scaffolds or natural ECM analogues [60-64].
Hepatocyte Growth Factor (HGF)
HGF, a well-known potent mitogen for endothelial cells that stimulates cell growth, is an attractive target for inducing angiogenesis [65]. In the phase III of clinical trial, intramuscular injection of plasmid DNA expressing human HGF was found to maintain limb perfusion, promote full ulcer healing rates, and reduce intermittent claudication syndrome in patients with CLI [66,67]. In addition, clinical studies conducted in Japan demonstrated that intramuscular injections plasmid encoding HGF tended to reduce pain and reduce ischemic ulcer size in patients with CLI [68,69].
An earlier preclinical study in rabbit models of HLI using a plasmid encoding the HGF728 and HGF723 isoforms showed that intramuscular injection of HGF effectively increased the number of collateral vessels and blood flow volume [70].
Angiogenic Factor With G-Patch and Forkhead-Associated Domain 1 (AGGF1)
Angiogenic factor that can effectively stimulate angiogenesis in animal models of HLI-angiogenic factor with G - patch and Forkhead - associated domain 1 (AGGF 1), was discovered during experimental studies. Intramuscular injection of a plasmid encoding human AGGF1 increased blood flow and vascular density induced by endothelial cells in ischemic hindlimbs of mice in an HLI model [71]. It was found that suppression of the generation of reactive oxygen species in the culture of endothelial cell precursors from animals with a diabetic model of HLI using AGGF 1 supported their function [72]. Activation of the integrin receptor for AGGF 1 on endothelial cells stimulated the cell signaling system for adhesion and migration and led to the formation of new capillaries [73].
Thrombin
Thrombin, as a key regulator of the hemostatic system, takes an active part in the progression of atherosclerosis. Thrombin belongs to the class of serine proteases. It is synthesized by a number of blood cells and the vascular endothelium in several stages, conforming from pre-prothrombin to prothrombin and thrombin. Thrombin precursor is secreted into the blood. The transition to thrombin is controlled by several factors of the blood coagulation system, including active serine peptidase factor X (Stewart–Prower factor F Xa), as a result of activation of factors III, VII, VIII, IX. The final transition to thrombin occurs with the participation of activated factor V (FVa), calcium ions and anionic phospholipids on the surface of platelets. As is known, during the formation of interendothelial leaks, platelet activation and the formation of their aggregates occur. Thrombin stimulates platelet aggregation through the direct participation of protease-activated platelet receptors (PAR-1,4) [74]. Thrombin takes an active part in all stages of the development of atherosclerotic plaque. The enzyme's application points are PAR receptors on platelets and vascular endothelial cells. Proteolysis of the intracellular N-amino-terminus of the receptor activates intracellular signaling pathways with the secretion of proatherogenic protein molecules: cyclooxygenase-2, MAP kinase, phospholipase C [75-77].
Migration of leukocytes from the vascular bed to the area of atherosclerotic plaque occurs upon contact with a family of proinflammatory cytokines, tumor necrosis factor-alpha, matrix chemoattractant protein-1, secreted in response to the presence of thrombin [78,79]. The presence of thrombin activates the vascular endothelium to express adhesion molecules (VCAM-1, ICAM- 1, von Willebrand factor), which support the process of active translation of leukocytes into the intimal zone of the ECM. The presence of thrombin in the ECM determines the translation of smooth muscle cells from the middle layer to the intimal layer with the formation of an atherogenic nucleus, accompanied by transformation into “foam cells” due to the accumulation of oxidized low-density lipoproteins in these cells [80]. The increased local inflammatory response is associated with platelet aggregation and the resulting expression of a large number of proatherogenic growth factors circulating in the ECM. In the presence of thrombin, hypervascularization of the atherogenic core occurs due to the active mobilization of the vascular endothelium with the release into the intimate zone of factors destabilizing the endothelial layer: angiopoietin-2 in conjunction with VEGF, FGF-2, MMP, PDGF. The progression of an atherogenic plaque is always accompanied by the recruitment and differentiation of pericytes during maturation and vascular remodeling both in the atherogenic core and beyond. Specific inhibition of thrombin activity in experiments leads to a decrease in atherogenic inflammation and suppression of plaque growth [81].
Interleukin Families (ILf)
Numerous mediators of vascular and systemic inflammation comprise the IL-1 family. This group includes several proinflammatory cytokines (IL-1α, IL-1β, IL-18, IL-33, IL- 36α, IL-36β and IL-36γ), as well as the IL-1 receptor (IL-1Ra, which support atherogenesis [82]. The formation of atherogenic inflammation is induced by several intracellular receptors (PRRs), and activation of PRRs accelerates the transition of IL-1β and IL- 18 precursors to active forms mediators of the IL -1 family and other proatherogenic families [83].
Interleukin-1 Beta (IL-1 β)
Interleukin-1b (IL-1b) is associated with acute and chronic inflammation. It is known that the level of IL-1b protein in patients with atherosclerosis is significantly increased compared to healthy subjects; the severity of the disease correlates with the level of IL-1b [84]. In the presence of a pro-inflammatory signal IL -1 b is synthesized by hydrolysis with the help of caspase-1 or neutrophil proteases from a precursor in monocytes and macrophages [85]. At the same time, protein recognition receptors (PRRs) are activated, IL-1a and IL-1b molecules bind to the IL-1 receptor (IL-1R), which forms an increasing cascade of inflammatory reaction under the vascular endothelium, leading to the massive uptake of cholesterol crystals and oxol by cells. LDL [82,86]. The binding of IL -1 b to IL -1 R 1 triggers signaling pathways for the expression of genes IL -1a, IL -1 b, IL -6, IL -8, MCP -1, COX -2 [87].
The inflammatory response in endothelial cells is manifested by the expression of intercellular inflammatory cytokine molecules (ICAM-1), vascular cell molecules (VCAM-1), and monocyte chemoattractant protein (MCP -1). Violation of the integrity of the endothelial layer is accompanied by increased secretion of IL-1b, intensive accumulation of cholesterol crystals and, in addition to macrophages, induction of proliferation of smooth muscle cells (SMS), their migration and transformation of the cytoplasm into a foamy structure, and expression of IL-6 [88]. The cocktail of inflammatory molecules supported by IL -1 b includes the products of cyclooxygenase metabolism of arachidonic acid, fibrinogen and plasminogen activator inhibitors, as well as a number of collagenases that can loosen and damage collagen structures by plaque fibrosis, starting with plaque remodeling (MMP - 3), further erosion of plaques (MMP -2, -9) and their rupture (MMP -1, -8, -13) [89-92].
The involvement of the direct effect of IL-1b on the formation and maturation of atherosclerotic plaques is experimentally proven by selectively switching off this cytokine using specific antibodies [93]. Neutralization of IL-1 β profiles the function of pro-inflammatory monocytes as reparative ones. This restructuring is accompanied by an increase in the level of anti-inflammatory cytokine IL -10 in the blood plasma and indicates a decrease in immune activation during atherogenesis in ApoE of mice [94].
Thus, a massive flow of cellular elements from the peripheral bloodstream and deep layers of the vessel wall is translated into the local intima with the participation of IL-1b [95]. Thus, the main mechanisms of participation IL-1β at the onset of atherogenic inflammation are endothelial dysfunction, monocyte migration and maturation, VSMC proliferation, IL-6 signaling to increase inflammation, increased MMP secretion [96].
Interleukin -1 Alpha (IL -1 α)
The key role of IL-1α in arterial remodeling during early experimental atherogenesis has been established [93,97]. IL-1α is primarily membrane bound and acts predominantly locally rather than systemically. This role of IL-1α in atherosclerotic experimental inflammation is confirmed using knockout of the IL-1 α receptor inhibitor molecule (IL-1 Ra) in C57BL/6J mice. Turning off the inhibitory IL-1 R a molecule during the development of atheroclerosis increased the levels of lipoproteins in plasma against the background of a cholesterol diet and caused the progression of cellular atherogenic infiltration in the vessel wall in the early stages [98]. The high activity of the IL -1 Ra inhibitor significantly reduces the degree of damage to the vessel wall by soft atherogenic plaques and affects the cellular and extracellular composition of atheroma [99].
Interleukin-4 (IL-4)
It has been established that IL-4 is present in high concentrations in atherosclerotic plaques in humans and mice, is involved in the esterification of oxidized LDL (oxLDL) and may play an important role in the dysfunction of vascular endothelial cells and the development of atherosclerosis, causing apoptosis of the vascular endothelium [100]. In the early stages of atherogenic inflammation, IL-4, against the background of oxidative stress, stimulates the secretion of cytokines and adhesion molecules such as VCAM-1, ICAM -1, IL-6, MCP-1, E- P - L -selectins in vascular endothelial cells. Adhesion to endothelial cells from the inside-out of monocytes, platelets and leukocytes, as well as cells of the T-system of immunity, determines the transendothelial migration of inflammatory reaction cells [101-103]. Next, the early cellular atherogenic reaction triggers the well-known chain of transformation of monocytes into macrophages and their massive capture of oxidized lipid mass through membrane scavenger receptors. Active proteinases destroy the intima and disinhibit the cellular composition of the middle layer of the vessel. The autoimmune reaction causes degradation of smooth muscle cells and plaque destruction [104]. A cascade of intracellular oxidative stress stimulates atherogenesis.
Interleukin-5 (IL-5)
Interleukin-5 (IL-5) is an antiatherogenic cytokine secreted by macrophages and mast cells, as well as representatives of the helper subpopulation of the T-immune system. The cytokine level increases with an increase in the titer of specific antibodies against ox-LDL in the blood plasma [105]. Experimental autoimmune suppression of IL-5 accelerates the development of atherosclerosi. [106] and, conversely, high expression of IL-5 increases the population of restored macrophages [107] and reduces the death of smooth muscle cells [108].
Interleukin-6 (IL-6)
The level of pro-inflammatory interleukin 6 (IL -6) is increased in atherosclerosis [109]. It is produced by macrophages, T lymphocytes and adipocytes, and is also a source of smooth muscle cells [110]. Increased levels of IL -1, TNF -α and interferon are accompanied by an increase in IL -6 levels [111]. Membrane- bound receptors for IL -6 (IL-6R) are expressed on neutrophils, monocytes, macrophages, and lymphocytes. In the acute phase of inflammation, proteins such as plasma amyloid A(SAA), C-reactive protein (CRP) provoke the start of atherogenic inflammation, activating platelet functions and the coagulation cascade. IL-6 induces monocyte translation through loss of endothelial layer integrity while upregulating the expression of adhesion molecules [96]. Increased synthesis of fibrinogen in the liver increases its infiltration in the intimal zone, the uptake of oxidized LDL and cholesterol crystals by macrophages [112].
Activation of IL -1 receptor antagonist (IL-1 RA) expression and release of soluble TNF -α receptor by IL -6 may lead to inhibition of both IL-1 and TNF-α activity [113]. This IL-6/ IL-6 R complex reacts on the cell surface with the universal gp 130, transsignaling leads to activation of inflammation [114].
Interleukin-7 (IL-7)
IL-7 is a pro-atherogenic cytokine that induces the activation of monocytes and the secretion of a number of inflammatory proteins by cells: IL -1 b, IL -8, MCP -1, as well as high expression in atherosclerotic plaques of a number of chemokine receptors: CCR 1, CCR 2, CCR 5 [115]. The action of IL -7 is carried out through affinity receptors (IL -7R) (CD127 and CD132), which have a similar structure of receptors IL-2, 4, 7, 9, 15 and 21. As a result of mitogenic activation of protein kinase, activation of NF- kB is triggered, which leads to the recruitment of monocytes and massive activation of macrophages [116].
Interleukin-8 (IL-8)
Interleukin 8 (IL-8) is a chemotactic factor, part of the beta- thromboglobulin superfamily, structurally related to platelet factor 4, attracts and activates neutrophils, basophils and T-cells during the inflammatory process, is expressed in response to inflammation by monocytes, macrophages, neutrophils. IL-8 is involved in mitogenesis, inhibition of angiogenesis, and induces neutrophil degranulation. The presence of IL -8 stimulates the proliferation and survival of endothelial cells and the production of MMPs [117].
Interleukin-9 (IL-9)
IL-9 has been demonstrated to aggravate the development of atherosclerosis in ApoE 2/2 mice, in part by inducing VCAM -1 expression and infiltrating inflammatory cells. Higher levels of IL-9 were found in plasma and carotid plaques in patients with carotid coronary atherosclerosis. The system of IL -9 and its specific receptor IL-9 R) on endothelial cells mediates the progression of atherogenic plaques in the mouse ApoE 2/2 model. At the same time, neutralization of IL -9 using monoclonal antibodies did not affect body weight or total plasma volume. cholesterol, triglycerides or high-density lipoprotein cholesterol [118].
Experimental knockout of anti- IL -9 using monoclonal antibodies with simultaneous injection of recombinant IL -9 (rIL -9) into mice significantly increased the lesion area and plaque size in the aorta and aortic root. At the same time, in the composition of atherogenic plaques, the number of macrophages with signs of high adhesiveness to the vascular endothelium increased due to the expression of vascular endothelial adhesion molecule-1 (VCAM-1), as well as T-cells. From the point of view of target cells, it is important that rIL -9 did not affect the functional properties of the Th1, Th2, Th17 and T-regulatory cell (Treg) subpopulations. The exclusive role of IL -9 in atherogenesis is also indicated by the stability of a number of proatherogenic and antiatherogenic cytokines, such as both in vitro and in vivo when introduced into the rIL -9 system (IL-4, IL-10, IL-17A, IFN-g).
Interleukin-10 (IL-10)
IL-10 family includes IL-10, IL-19, IL-20, IL-22, IL-24, IL-26, IL- 28A, IL-28B, and IL-29. Members of the family play a critical role in suppressing atherogenic inflammation. Interleukin (IL)-10 is an anti-inflammatory cytokine produced primarily by macrophages and Th2 T-lymphocytes. Convincing evidence has been provided that the levels of IL-10 in the blood plasma in the late stages of experimental atherosclerosis increase compared to the control group, provided that IL-1β is neutralized, while IL-10 has a lipid- lowering effect, since the level of cholesterol in the blood serum is significantly decreases in mice treated with IL-10 [93,119].
IL-10 has been shown to be produced by IL-1β - deficient macrophages, inhibit MHC class II expression, and recruit reparative monocytes to reduce atherogenic inflammation [94,120]. The secretion of molecules of the IL-10 family suppresses oxidative stress and the adhesion of monocytes through the vascular endothelium, increases the uptake of lipids by macrophages and reverse cholesterol transport, while reducing the deposition of cholesterol esters, reduces the activity of cellular proteinases and proteins of the IL -1 family, interfering with cyclooxygenase the arachidonic acid metabolic pathway, as well as increasing the translation of cholesterol into the ECM [96,121].
Interleukin -11 (IL-11)
Interleukin-11, a mature secreted protein consisting of 178 amino acids with a molecular weight of approximately 20 kDa, is a member of the IL-6 family of cytokines and directly affects vascular function, including IL-6 and IL-27 [122]. In healthy people, the expression of IL-11 in the blood plasma is practically undetectable; low level of IL -11 is found in the absence of inflammation in endothelial cells, smooth muscle cells and fibroblasts of the vascular wall. IL-11 expression is significantly altered by TGF-β and IL-1 stimulation. [123]. IL-11 signaling involves the interaction of the soluble form of IL-11 with the IL-11RA and GP130 receptors in the cell to form a trimeric complex. IL-11Ra expression occurs in endothelial cells, smooth muscle cells and fibroblasts during the development of ischemia and inflammation [124-126]. The interaction of IL-11 with its receptor triggers the expression of glycoprotein 130 (GP 130) in neighboring cells, which leads to signal transmission [127]. It has been established that IL -11 remodels the function of endothelial cells, smooth muscle cells and fibroblasts in the vascular wall [128]. It is important that in cultured human umbilical vein endothelial cells, the expression of VEGF triggers overexpression of IL-11. During the remodeling of endothelial cells, IL -11 as a recombinant molecule is able to prevent endothelial destruction when introduced into the system of cytotoxic lymphocytes [129]. Remodeling of VSMCs in the presence of IL -11 can be inhibited if the system suppresses the signal for cytokine expression, despite the initiation of TGF-β and Ang-2 [130]. Neutralization of IL- 11 using a monoclonal antibody reduces not only the number of VSMCS, but also the level of matrix metalloproteinase 2 (MMP2) and collagen content. This maintains the contractile phenotype of VSMCs. In a model associated with plaque formation, compared with the control group, knocking out IL-11 significantly reduced vascular wall thickness and atherogenic damage. In the vascular adventitia layer, the secretion of IL -11 can be induced by TGF-β, which results in activation of fibroblasts and an increase in the inflammatory response [131].
Interleukin-12 Family (IL-12f)
Members of the interleukin-12 family (IL-12, IL -23, IL-27, and IL-35) are a class of cytokines that regulate various biological effects, and their activity is associated with the progression of atherosclerosis [132]. Interleukin-12 (IL-12) is secreted by monocytes, macrophages, neutrophils, dendritic cells, lymphocytes of the T-immune system Activation of IL-12 secretion by cells can be triggered by polysaccharide components of the ECM, stimulating the adhesion and translocation of inflammatory cells in atherogenic plaques at the early stage of atherogenesis in an experimental model of hypercholesterolemia. IL-35 is mainly secreted by T-helper cells [133].
IL-12 and IL-23 are considered pro-inflammatory factors that enhance downstream inflammatory signals. Exogenous experimental administration of recombinant murine IL-12 to mice leads to the progression of atherosclerosis, increases the area of atherosclerotic plaques in the aorta, as well as when using ApoEknockout mice and with deficiency of low-density lipoprotein receptors (LDL). Abrogation of the biological effect of IL-12 significantly reduces cardiac dysfunction in animals and promotes angiogenesis [134].
Interleukin-13 (IL-13)
Interleukin 13 is a protein secreted by T-helper type 2 (Th2) cells, NK cells, mast cells, basophils and eosinophils. Exogenous administration of IL-13 favorably altered the morphology of established atherosclerotic lesions, increased plaque collagen content, and decreased the secretion of vascular cell adhesion molecules VCAM-1. As a result of the anti-inflammatory effect, a decrease in the number of macrophages is recorded in plaques due to decreased recruitment and adhesion of monocytes in the affected vascular wall. Moreover, under conditions in in vitro clearance increases o xLDL as a result of reformatting of activated macrophages (M1) into alternatively anti-inflammatory macrophages (M2). This effect indicates protection against atherosclerosis by IL-13. Protection is selectively mediated by signaling through the IL-13 receptor (IL-13Ra2), which induces TGF-b1 production in macrophages and increases the number of anti-inflammatory macrophages [135,136].
In a study, exogenous administration of IL-13 to LDLR /mice for 4 months did not reduce the number and size of atherogenic plaques compared to control (PBS), despite a threefold increase in the dose of IL -13, and also showed no changes in morphological analysis [137]. However, collagen content was significantly higher in atherosclerotic LDLR/mice treated with intraperitoneal IL- 13 compared with PBS-treated mice, suggesting macrophage or smooth muscle cell stimulation of collagen secretion.
A targeted study of the content of the culture of “foam” cells upon administration of IL-13, pre-loaded with cholesterol, showed a very high cholesterol content in the cytoplasm, which is regarded as an active outflow of cholesterol from the ECM into this population of macrophages. This clearance is considered to effectively remove pro-inflammatory OxLDL and reduce the severity of the inflammatory response. IL-13 deficiency generated in various mouse models revealed significantly accelerated atherosclerosis with almost twice as large aortic lesions and increased necrotic core formation, consistent with plaque progression.
Interleukin-15 (IL-15)
Interleukin-15 (IL-15) is a cytokine belonging to the interleukin-2 (IL-2) family, has general biological effects, including immunoregulation through and induction of natural killer cell proliferation [138]. The protein is secreted by phagocytes and mononuclear cells. It is known that as atherosclerosis progresses, the level of IL-15 increases. A number of studies on mouse models of myocardial infarction or myocarditis have found a protective effect manifested in the preservation of viable cardiomyocytes [139].
Interleukin-17 (IL-17)
Interleukin-17 is a member of a family of variably homologous proteins (IL-17A- F). IL-17A has a pro-inflammatory effect and interacts with such mediators as IL-1β, IFN-γ, GM-CSF, IL-22, TNF-α [140]. IL-17 acts independently or synergistically with additional proinflammatory mediators, stimulating the production of chemokines [141], granulocyte-colony-stimulating factor (G-CSF). IL-17A stimulates the release of IL-2 from Th cells, which in turn increases the number of regulatory T-cells [142]. In addition to the chemokine family, IL-17 can induce a number of antimicrobial peptides, including inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX -2) [143]. The action of IL- 17 is carried out through five cytokine receptors (IL-17 RA-E), among which IL -17 RA is a common receptor for various forms. It has been established that cholesterol and oxLDL stimulate the secretion of IL-17 directly and indirectly through cytokines; its expression increases significantly with the progression of atherosclerosis. The activity of Th 17 cells, which express not only IL-17, but also a number of its structural analogs (IL-17A, IL-17F), as well as TNF -α and IL-22, depends on the state of cellular and intercellular lipid metabolism [144]. Despite the lack of direct evidence of the pro-atherogenic effect of IL-17 in animal models of atherosclerosis, attention should be paid to the work [145], in which the authors used the introduction of recombinant IL -17A into Ldlr −/− mice and discovered a direct role for IL-17A in modulating atherosclerosis. The authors are inclined to believe that neutralization of IL-17A stimulates the pro-atherogenic Th1 pathway, which may be responsible for increased atherogenesis [146]. In addition, this version is supported by the ability of IL- 17A to suppress the expression of inflammatory adhesion molecule (VCAM-1). The atheroprotective role of IL-17 was demonstrated in works [147,148]. However, most studies indicate a synergistic proinflammatory effect of IL-17, IFN-g secreted by T-cells and smooth muscle cells of the vascular wall with the participation of positive regulators IL-21 and IL-23. This synergy is associated with the progression of inflammation in atherogenic plaques [149,150].
Interleukin-18 (IL-18)
IL -18 can determine endothelial cell dysfunction, enhance monocyte migration and maturation, induce VSMC proliferation, potentiate the synthesis of IL-1α, IL-1β, enhance the inflammatory process through IL -6 signal transduction, increase production MMP, and, consequently, promote the progression and degradation of plaques, enhance the secretion of TNF-α, IFN-γ by macrophages and smooth muscle cells [151-153].
Interleukin-19 (IL-19)
IL -19 is a member of the IL -10 subfamily and exhibits anti- inflammatory effects [154]. Studies have revealed that in the wall of healthy arteries, the expression of IL-19 is practically absent and is actively manifested in atherosclerotic plaque. Systemic use of the recombinant IL -19 molecule in LDLR −/− mice against the background of an atherogenic diet compared to the control is antiatherogenic, reduces macrophage infiltration in atherosclerotic lesions, reduces the interaction of leukocytes with the vascular endothelium in vivo, significantly reduces the adhesion of leukocytes and monocytes to monolayers of endothelial cells in vitro, pre-stimulated with oxLDL, reduces the area of damage to the main artery. Efferocytosis of apoptotic cells and profiling of pro-atherogenic M1 macrophages into reparative M2 macrophages are characteristic of systemic administration of IL -19 to the experimental system, which corresponds to regression of atherogenic inflammation [155].
Thus, plaque regression results from modulation of cholesterol transport and M2 macrophage polarization [156-158]. Pre-treatment of macrophages with IL-19 and subsequent introduction of fluorescently labeled oxLDL into the system shows that the volume of lipid uptake increases approximately 1.5 times. The presence of increased levels of IL-19 in the system is accompanied by the expression of CD36, SRA-1 and SRB-1 molecules, which are key participants in the uptake of modified lipids by macrophages and have atheroprotective properties [159]. Cholesterol accumulation in macrophages is directly related to CD36 expression [160]. Overexpression of SRA-1 in ApoE knockout mice reduces the level of atherosclerotic lesions [161]. The same overexpression of SRB-1 is able to protect against atherogenic damage in mice with LDLR [162].
The transition of macrophages from type M1 to type M2 is accompanied by an increase in the expression of scavenger receptors in cells [163]. Recruitment of macrophages means reverse transport of accumulated cholesterol. M acrophages export excess cholesterol to apolipoprotein A1 (apoA1) and high-density lipoprotein. This ensures reverse transport of cholesterol to the liver [159].
Reduced inflammation is accompanied by decreased cell adhesion with decreased VCAM1 immunoreactivity [155,164]. Similar results were obtained using ApoE −/− mice. These cellular dynamics were accompanied by a decrease in the levels of potent proinflammatory cytokines mRNA for IL-1β and IL-12p40, IFNγ, IL-8 and MCP-1. Despite a significant reduction in the area of local damage to the vessel wall by atherogenic plaques with systemic administration of IL-19, plasma lipid levels did not change compared to the control. This indicates the absence of a lipid-lowering mechanism of action IL-19. The same expression profiles by macrophages treated with TNF-a have been reported in patients with BMDM. In this case, the macrophage line of the M2 phenotype, unlike M1, expressed IL -19. Importantly, the antiatherogenic effect of the cytokine induces beneficial polarization of T-cells, changing the Th1 to Th2 phenotype.
Interleukin-20 (IL-20)
Interleukin-20 is a family of cytokines IL-19, IL-20, IL-22, IL-24 that transmit their signals through the receptor β chain of IL-20 receptor complexes (IL-20Rα, IL-20Rβ) Types I and II [165,166]. Activation of the receptor complex triggers the downstream signaling system of Janus kinase (JAK) and the signal transduction and activator of transcription (STAT) pathway [167]. This IL-20 system is realized in resident endothelial cells of the intima and smooth muscle cells of the middle layer of the vessel.
The cytokine IL-20 is found in the endothelium and macrophages of the atherogenic plaque, but remains intact when oxLDL is taken up by macrophages [168]. Researchers associate the proatherogenic nature of IL-20 with the activation of VEGF in vascular endothelial cells and the angiogenic effect in the form of morphological reconstruction of new vessels [169].
Interleukin-22 (IL-22)
IL-22 is a member of the IL-10 family of cytokines and is secreted by activated T-cells, especially T-helper (Th22) cells and Th 17 cells, NK cells, neutrophils, macrophages, fibroblasts, smooth muscle cells (VSMC) of unstable atherosclerotic plaques [170,171].
Most studies point to a proatherogenic mechanism of action for IL-22. Cytokine regulates proliferation and migration VSMC from the middle layer of the vascular wall into the intimal layer of the main artery enhances the inflammatory response in the form of accumulation of macrophages and neutrophils, stimulating the secretion of pro-inflammatory chemokines. The action of the cytokine is aimed at differentiating macrophages from an anti-inflammatory to a pro-inflammatory phenotype and reducing the ability of cells to translate cholesterol, which increases the formation of cells with foamy cytoplasm [172,173]. The start of the inflammatory reaction on the part of endothelial cells induces, along with other cytokines, IL-22, regulating the expression of adhesion molecules ICAM 1 and VCAM-1 [174]. Knockout of IL-22 from a mouse experimental system leads to a decrease in plaque size [175]. 12 weeks of feeding a cholesterol diet to Apoe−/− mice with simultaneous administration of recombinant murine IL-22 (rIL-22) three times a week led to an increase in the size of atherosclerotic plaques in the aortic root and in the aorta itself compared with control Apoe −/− animals who received PBS. Treatment of animals with 20 μg of monoclonal antibody against IL-22 (IL-22 mAb) proved the causal role of the cytokine in the formation of atherosclerotic inflammation.
Thus, the study results indicate that IL-22 plays a key role in atherosclerotic plaque formation by modulating the contractile pattern of VSMCS towards synthetic repair cells. As a result of this regulation, the content of elastin and collagen in the ECM increases, which thickens the fibrous cap of the atherogenic core and increases the stability of the plaque. Despite the increase in the mass of foamy macrophages in the plaque and its fibrous strengthening, the process means, one way or another, the progression of atherosclerosis [174].
Interleukin-23 (IL-23)
IL-23 is secreted by macrophages and dendritic cells; the target cells for the cytokine are mainly Th17 lymphocytes. Administration of IL-23 to experimental mouse models of atherosclerosis reduced inflammatory responses [176]. However, other studies indicate a proatherogenic function of this cytokine [177].
Despite conflicting research results on the role of IL -23 in atherosclerosis, the proinflammatory effects of IL -23 may be mediated through other growth factors, such as GM-CSF or cytokines, such as IL-22 [176,178-180].
Interleukin-24 (IL-24)
IL-24 is able to induce the secretion of TNFα and IL-6 by monocytes, which could potentially exhibit a pro-atherosclerotic effect. On the other hand, inhibition of the production of reactive oxygen species in cultured smooth muscle cells reduces their proliferation and, thereby, reduces atherogenic inflammation. The dual function of this cytokine requires specific activation or inhibition studies.
One study using cultured human VSMCs suggests that IL-24 inhibits the production of reactive oxygen species (ROS), thereby reducing ROS-induced VSMC proliferation, a major maladaptive event in atherosclerosis [181]. Taken together, IL-24 can have both pro- and atheroprotective effects, which poses the task of targeted elucidation of the function using animal models of cytokine activation or incorporation.
IL-25 is a member of the IL-17 cytokine family, stimulates the expression of IL-4, IL-5 and IL-13, regulates the Th2-dependent immune response, which confirms the atheroprotective profile of the immune response of Th 2 cells in models of experimental atherosclerosis [182]. IL-25 is expressed by endothelial cells, macrophages and intimal T-cells [183]. Exogenous total or temporary deficiency of IL-25 in double knockout mice (Apoe-/- IL-25-/-), as well as against the background of a high cholesterol diet, changes the expression profile of T-cells towards Th1/Th17 and ensures the progression of atherosclerosis in the form an increase in the area of plaques in the aortic arch. The secretion of proatherogenic cytokines in the form of IL-1, IL-6, 12(p40), TNFα, as well as IFN-γ confirms the worsening of atherogenic damage to the great vessel [184]. Exogenous deficiency of IL-25 leads to a decrease in the amount of IgM antibodies in plasma that bind oxidized LDL [185]. However, the complete absence of IL -25 in blood plasma does not change its lipid profile.
Interleukin-27 (IL-27)
IL-27 is an anti-inflammatory cytokine with a broad spectrum of activity [186]. The targets of the cytokine are the vascular endothelium and all hematopoietic cells. IL-27 suppresses CD4+ T cell activation. IL-27 receptor deficiency demonstrates the accumulation and activation of Th1 and Th17 CD4+ T-cells in the wall of the main artery, increased levels of IL-17A, and infiltration of inflammatory cells [187]. IL-27 reduces LDL content in macrophages and foam cell formation [188].
Interleukin-32 (IL-32)
IL-32 is a proinflammatory cytokine produced and released from both immune cells (NK cells, macrophages, monocytes and T-lymphocytes, including regulatory T lymphocytes, and non- immune cells, including endothelial cells [189-192]. However, in most cases, IL -32 is found mainly intracellularly, e.g. IL-32β in the endoplasmic reticulum (ER) of human endothelial cells, but can change localization depending on the state of the cells [193].
The cytokine is expressed in at least 10 different isoforms (α, β, γ, δ, D, ε, θ, ζ, η and small / sm, generated by alternative splicing) [194,195]. It has been observed that a person with signs of obesity has increased levels of circulating IL-32. It has been established that the endothelium of the coronary arteries in people with coronary artery disease significantly expresses IL-32 [196,197]. High expression of IL-32 β and IL-32 γ mRNA was found in the atherosclerotic wall of human arterial vessels, and more precisely, in M 1/ M 2 macrophages [198].
Regulation of ICAM-1 and VCAM-1 molecules on the vascular endothelium promotes strong adhesion and migration of monocytes across the endothelium. Subsequent overexpression of IL-32γ in macrophages leads to increased production of a number of chemokines, as well as cell adhesion molecules sVCAM-1, interstitial collagenase (MMP-1), gelatinase (MMP- 9) and collagenase 3 (MMP-13), leading to the recruitment of vascular endothelium, masses of monocytes and macrophages, smooth muscle cells of the middle layer of the vascular wall with a significant decrease in collagen synthesis and destabilization of atherogenic plaques Knockdown of IL-32 resulted in a decrease in both constitutive and IL-1β induced intercellular adhesion molecule-1 (ICAM-1), IL-1α, IL-6, IL-8 [192]. Numerous studies show that IL-32 affects cholesterol levels by interfering with efflux and transport of HDL particles.
IL-32 triggers the induction of a number of pro-inflammatory cytokines TNF α, IL-1β, induced, in turn, by ICAM-1, IL-6 and IL- 8, which leads to the progression of the formation of atherogenic plaques [190,192,199,200] .The secretion of IL-1, IL-12, IL-18 and TNF-α, in turn, maintains the expression of IL-32 [199]. Increased expression of IL-32 in M2 type macrophages is due not only to the presence of in in vitro IFNγ, but also as a result of stimulation by a combination of IFNγ and TNFα.
Interleukin-33 (IL-33)
It is assumed that IL-33 has atheroprotective properties, increases the permeability of the endothelial barrier and activates angiogenesis, initially stimulating the expression of endothelial adhesion molecules VCAM1, ICAM1 and E-selectin, which leads to increased leukocyte adhesion [201,202]. The cytokine is able to increase the production of natural IgM antibodies against oxLDL foam cells of macrophages and prevents their formation [203,204]. When regulating atherogenic inflammation, IL-33 is expressed by macrophages, endothelial cells, dendritic cells, and fibroblasts [136]. The cytokine activates type 2 innate lymphoid cells, increases the polarization of macrophages towards the M2 phenotype, inhibits the formation of foam cells, enhances the polarization of T-cells from Th 1 to Th 2, and activates the function of T-regulatory cells [96,205].
Interleukin-35 (IL-35)
This anti-inflammatory cytokine is produced by T-regulatory lymphocytes (Treg) and B cells. IL-35 has an anti-inflammatory effect and protects against tissue damage during the inflammatory response [206]. The target cells of IL-35 are Tregs themselves, Th2 lymphocytes, endothelial cells, monocytes, smooth muscle cells of the middle layer of the vascular wall. For example, the striking anti-inflammatory effect of IL-35 can be demonstrated by inhibiting the expression of VCAM-1 by endothelial cells in a model of LPS -induced acute inflammation by inactivating the mitogen-activated protein kinase (MAPK) signaling pathway [207]. Exogenous administration of IL-35 allows, in a mouse model of myocardial infarction, to significantly reduce the affected area of the left ventricle, reduce the death of macrophages, and increase the differentiation and activity of M2 cells [208]. In a rat model of coronary artery disease, IL-35 treatment significantly promotes early endothelialization of drug-eluting stents. These studies demonstrate that IL-35 regulates the differentiation of various immune cells involved in the progression of atherosclerotic heart disease.
In conditions in In vitro treatment of monocytes with inflammatory mediators IL-1b, IL-18, IFNγ, TNFα, LPS produces a response in the form of IL-37 expression [212]. In conditions in vivo, the same pattern is confirmed in a rabbit model of atherosclerosis when analyzing foam cells of atherogenic plaques. In the apoE − / − mouse model, injections of recombinant IL-37 for 4 months resulted in decreased atherosclerotic plaque size and increased stability, decreased serum levels of proinflammatory cytokines, and decreased vascular calcification compared to control mice [213]. Creation of exogenous IL-37 deficiency using antibodies leads to a reversal of the anti-inflammatory effect [211]. Intracellular cytokine staining technology has shown that, in addition to macrophages, infiltrated CD4+ T lymphocytes and vascular smooth muscle cells (SMCs) in areas of calcified plaques are the main sources of IL-37.
Tumor Necrosis Factor-Alpha (TNFα)
The tumor necrosis factor superfamily (TNFSF) plays a role in both the protection and progression of atherosclerosis [214]. The most important role in the development of atherosclerosis is played by TNF -α, TNF- related apoptosis-inducing ligand (TRAIL), TNF like apoptosis inducer (TWEAK), CD40L and related receptors. The induction of pro-apoptotic and cell-protective general signaling pathways occurs through the TNF-R1 and TNF-R2 receptors. [215,216].
Tumor necrosis factor-alpha is a pro-atherogenic cytokine at sufficiently high levels in plasma, capable of causing a pro- angiogenic effect by stimulating the expression of cytokines, cell adhesion molecules [217], migration and mitogenesis of endothelial cells and smooth muscle cells in the vascular wall, causing the expression of macrophage colony-stimulating factor (M-CSF) with subsequent proliferation and differentiation of monocytes, survival and maturation of macrophages [218,219]. In this case, two processes occur simultaneously: the formation of new microcapillaries and increased formation of atherogenic plaques. TNFα is produced by monocytes and macrophages, and its involvement in the pathogenesis of atherosclerosis is confirmed by its presence in human atherosclerotic plaques.
Experimental knockout of the TNF - α system in hyperlipidemic mice reduces atherogenesis, endothelial adhesion, and levels of inflammatory markers compared to controls [220]. This effect of blocking TNF-α is accompanied by an increase in high- density lipoprotein cholesterol. On the contrary, high levels of TNF-α disrupt the mechanism of normal reverse cholesterol transport, enhance endothelial dysfunction and oxidative stress, induce transmigration of monocytes into the subendothelial space, and increase the uptake of oxLDL by newly formed M1 macrophages when activating the expression of class A scavenger receptors [221]. The effect is completed by the retention of LDL in the subendothelial space, increased cell apoptosis by induced macrophages, and translocation of VSMC towards the accumulation of macrophage mass [96].
Interferon-α (INF-α)
Administration of IFN-α has a proatherogenic effect and accelerates the development of atherosclerosis [222]. This antiangiogenic effect of type I IFN is thought to be primarily due to their antiproliferative activity against EC [223]. IFN -α includes about 20 structurally related proteins, which are mainly produced by monocytes and plasma cells, precursors of dendritic cells (PDC). Upon short-term exposure, INF-α suppresses endothelial cell apoptosis [224]. Long-term exposure of the cytokine to cells, which is typical in chronic atherogenic inflammation, in the presence of growth factors induces increased adhesion of mononuclear blood cells to the endothelial lining, which accelerates the development of atherosclerosis.
The results of studies by other authors partially contradict previous results and indicate antiproliferative effects of the cytokine, as well as, moreover, its pro-apoptotic activity [225-229].
Interferon-γ (IFN-γ)
IFN-γ is a proatherogenic cytokine secreted by activated T lymphocytes in the atherogenic plaque. Transmission of the IFN-γ signal into the cell occurs by interacting through a heterodimeric cell surface receptor consisting of two separate subunits: IFN-γ receptor (IFNGR)-1 and IFNGR2 [230].
The cytokine is able to increase the activity of endothelial cells, enhance the mobilization of polymorphonuclear white blood cells, induce macrophages towards the atherogenic M1 phenotype, enhance the polarization of T-cells towards the Th1 phenotype, reduce collagen synthesis by smooth muscle cells, disrupting plaque stability [96,231,232]. The process of transformation of macrophages into the M1 phenotype is accompanied by a decrease in cholesterol synthesis, accumulation of cholesteryl esters in lipid droplets, and stimulation of the production of cholesterol derivatives [233]. As a result, the phagocytic ability of macrophages to capture lipids increases. This induces the secretion of other atherogenic cytokines, such as IL-1β, IL-6, TNFα, IL- 12, IL-18, enhancing the secretion of IFN- γ by macrophages and T-cells, chronic inflammation. The synergistic interaction of proatherogenic cytokines significantly enhances the inflammatory response [234]. However, no changes in serum cholesterol levels were observed.
The proatherogenic effect of IFN -γ is confirmed in the IFN -γ receptor-deficient Apoe −/− mouse model. Low levels of the cytokine are accompanied by a decrease in plaque lipid mass and inflammatory cell infiltration [235].
Transforming Growth Factor-a (TGF-a)
Transforming growth factor-alpha (TGF-β) has attitude to induction structures of extracellular matrix [31]. TGF-ðÂÃÃÃÂ???ÂÃÃÂ??ÂÃÂ?ÂÂ?¼ is a member of the epidermal growth factor (EGF) family, single-chain peptides of 50- 53 amino acids cross-linked by three disulfide bonds. The cytokine activates transmembrane tyrosine kinase EGF receptor (EGF-R), increases intracellular calcium levels, glycolysis and EGFR gene expression. The interactions of TGF-alpha and TGF-beta can be either synergistic or antagonistic.
Transforming Growth Factor-β (TGF-β)
Transforming growth factors β (TGF-β) are a superfamily of proteins containing approximately 20 members of the bone morphogenetic protein (BMP) family. The family of proteins plays an important role in maintaining tissue homeostasis, in particular in the processes of vascular remodeling by inducing intracellular signals through serine/threonine kinase receptor subtypes I and II. TGF-β1 plays an important role in inflammatory processes and is found in endothelial cells, macrophages, smooth muscle cells of the middle layer of blood vessels, and myofibroblasts [236]. The cytokine is found in plasma and binds to extracellular matrix proteins, induces increased expression of adhesion molecules ICAM-1 and VCAM-1, remodels the extracellular matrix, stimulates fibrogenesis, regulates the recruitment of leukocytes and fibroblasts [237,238]. The cytokine is involved in the growth, development and proliferation of smooth muscle cells and fibroblasts [239]. In a knockout (Apoe–/–) mouse model, the presence of TGFβ induces the recruitment of M1 macrophages to the site of inflammation. However, in close interaction with T-regulatory (Treg) cells with known immunomodulatory activity, mediated secretion of anti-inflammatory and atheroprotective cytokines, including IL-10 and IL-35, plays an important role in the regression of atherosclerotic plaque [240]. Overexpression of the cytokine reduces the risk of atherosclerosis and, conversely, inhibition of TGF -β signaling promotes the development of atherosclerosis [241].
TGF-β family includes growth differentiation factor-15 (GDF- 15), as a cytokine that inhibits M1 macrophages. The cytokine is expressed in endothelial and smooth muscle cells, macrophages, and adipocytes under normal conditions and during inflammation [242]. Many proatherogenic and antiatherogenic cytokines are capable of inducing GDF-15 expression [243]. GDF-15 increases the vulnerability of plaques in the early stages of atherosclerosis due to pro-inflammatory and angiogenic effects [244,245].
Platelet-Derived Growth Factor-BB (PDGF-BB)
PDGF is a family of closely related small molecular weight proteins, consisting of five isoforms: PDGF-AA, PDGF-AB, PDGF-BB, PDGF-CC and PDGF-DD, polypeptides linked by disulfide bonds [246]. Subtypes of PDGF-A and B are activated inside cells, subtypes PDGF-C and D are secreted in an inactive form and require activation by extracellular proteases [247-249]. It has been reliably shown that PDGF is involved in the maturation and remodeling of blood vessels and affects ECM parietal cells [250,251]. These growth factor isoforms are required for pericyte recruitment and differentiation during vessel formation [252]. Recruitment of pericytes by PDGF is carried out through two receptor tyrosine kinases, PDGF α and β [253,254]. This recruitment of pericytes and smooth muscle cells as a result of PDGF adhesion in the ECM environment occurs due to the strong binding of positively charged amino acids through the carboxy- terminal region [255]. Studies have found that the transformative effectiveness of PDGF - D is similar to that of B and C, the use of PDGF - C led to an increase in the number vessels, PDGF-B and D stimulated the formation of larger vessels [256].
It has been established that the PDGF signaling pathway is enhanced by the action of FGF on EC. At the same time, FGF increases the expression of PDGF-B and D, which is very important for the recruitment of perivascular cells and remodeling of the vascular system [257]. Maturation of endothelial tubules and blood perfusion induces vascular endothelium to release platelet-derived growth factor [258,259]. PDGF recruits mural cells through expression of the PDGF receptor R and induces their differentiation into pericytes and smooth muscle cells. Next, opsonization of endothelial tubules by these cells occurs, which ends with the formation of stabilized vessels [260,261].
An important role in the regulation of angiogenesis is confirmed by the fact that the removal of the influence of PDGF on the vascular wall leads to the removal of the effect of proliferation and migration of smooth muscle cells of the middle layer of the artery (VSMCS) [262]. At the same time, the regulatory role of VEGF remains. For example, pretreatment of ECS with VEGF for 8 h results in upregulation of PDGF-BB, PDGF-DD, and heparin- binding epidermal growth factor, which play a role in pericyte proliferation and recruitment [263]. Due to the formation of new intercellular contacts between endothelial cells and pericytes, the final stabilization of the vasculature and restoration of the ECM is accompanied by the expression of transforming growth factor-β (TGF-β) with the participation of endothelial cell adhesion molecules [264].
The use of several variants of the polypeptide chains of this growth factor in the free state or as part of an adeno-associated plasmid revealed an angiogenic effect in various models of ischemic vascular diseases, in particular in hind limb ischemia in diabetic mice [265]. Experimental regulation of the level of PDGF activity confirmed the angiogenic effect of this growth factor.
It is important to note that high concentrations of PDGF in the vascular wall mean excessive proliferation of cells of the mural system with disorganization of the smooth muscle layer and pericytes, which is characteristic, for example, of the activation of atherogenesis [266]. Thus, local determination of the level of this growth factor is important for comparative analysis with the activity of other polypeptide substances.
Matrix Metalloproteinases (MMP-2, MMP-9)
During in vivo angiogenesis, MMPs degrade type IV collagen in the ECM as a major component of subendothelial basement membranes and are associated with endothelial cell migration and pericellular fibrinolysis. The process of cleavage of intercellular adhesion exposes binding sites and induces the release of ECM- associated angiogenic growth factors. This ends with the formation of new blood vessels [267,268]. The state of pericytes in the ECM has been shown to influence MMP activity. Impaired mobilization of pericytes due to MMP deficiency is accompanied by blocking the detachment of pericytes from the endothelial layer, which significantly reduces the migration of endothelial cells during the period of proliferation into niches that should be formed during pericyte detachment [269]. This condition indicates the completion of remodeling of the vessel wall in the area of inflammation and the restored stability of the vascular system [270]. If you create dual delivery of several growth factors using matrices, for example, VEGF, FGF-2 and PDGF, then the expression of MMP (MMP -2) increases significantly. Loose artificial structures do not create this effect.
Remodeling of The Basement Membrane During Ischemia Remodeling of the basement membrane and growth of the vascular network under the influence of angiogenic factors is expressed in the destruction of the basement membrane, detachment of pericytes from the basement membrane, which allows endothelial cells to expand the migration zone and occupy the vacated niche [17,56,271]. Initially, at rest, pericytes are found in the microvasculature, expand their processes and envelop the endothelial monolayer in the capillaries. To identify pericytes, surface markers (platelet-derived growth factor receptor, PDGFR)-β, CD13, cytoskeletal proteins (α-smooth muscle actin, α-SMA, desmin and calponin) are determined [272,273].
The stabilization process ends with the compaction of interendothelial contacts and a decrease in the migratory activity of endothelial cells [274]. The inclusion of tissue inhibitors of metalloproteinases (TIMPs) and plasminogen activator inhibitor-1 (PAI -1) in the process promotes reconstruction of the basement membrane [275,276].
The composition of the formed basil membrane includes laminin isoforms, type IV collagen, perlecan, nidogens, fibronectin, proteinase inhibitor [277-282]. During ischemia and inflammation, the ECS adhesively interacts with the basement membrane scaffold through key integrins on the EC surface, including fibronectin receptors (FN), α5β1, laminin receptors [LM], α6β1 and α3β1, collagen receptor (K), α2β1, and arginine binding receptor- glycine–aspartic acid (RGD), av β3). This complex adhesion of endothelial cells allows interaction with many ECM proteins.
The intimate layer of the ECM is a proteoglycan-rich matrix that includes interstitial collagens of various types and adhesive glycoproteins. The middle layer of the artery is a complex of glycoprotein elastic plates, microfibrils, which controls the differentiation of smooth muscle cell functions [283,284]. The mechanical strength of the middle layer of the artery to the stresses of blood flow is ensured by cross-linking of the matrix with transglutaminases. The construction of the ECM of the adventitial layer indicates its readiness for activation after vessel injury [285- 288].
In addition to the leakage of plasma components during ischemia and inflammation into the intimal space due to increased vascular permeability from the capillaries of the vascular bed, there is a leakage of cellular composition and growth factors into the adventitial space. Due to this translation mechanism, the adventitial vasculature expands with leaky vessels as atherosclerosis progresses [289-291].
Cells of the inflammatory reaction penetrate into the walls of blood vessels on both sides, which is very important when translating hydrogel constructs into the paraadventitial space in the case of solving the problem of modeling a new vascular network [292- 294].
Excessive growth of cellularity of the intimal layer develops not only as a result of an imbalance of concentric elastic plates, proliferation, invasion and loss of VSMC from the middle layer of the great vessel, but also as a result of the active inclusion of proliferating pericytes in contact with leukocytes, macrophages, lymphocytes, mast cells and fibroblasts with the participation cocktail of growth factor-associated proteins (LTBP-1, latent TGFβ-binding protein 1), IGFBP-3 (insulin-like growth factor binding protein 3), CTGF (connective tissue growth factor), gremlin, follistatins), fibrillin- 1, fibulins, FN) and the growth factors themselves, emanating from the capillary vascular network of the adventitial zone: TGFβ1 - transforming growth factor, VEGF - vascular endothelial growth factor, HB-EGF - heparin-binding epidermal growth factor [295,296]. As a result of remodeling of the middle layer of the ECM, invasion of vascular cells into the intimal space from the middle layer, as well as from the adventitial space into the middle layer, is realized. In addition to cellular infiltration into the expanded intimal space in atherosclerosis, deposition of proteoglycans and interstitial collagen occurs [297]. As a result of the inflammatory reaction, the formation of defects in the endothelial layer is compensated by fibrin synthesis and platelet activation, which stimulates the release of PDGFs and TGFβ1, as well as a number of mediators (thrombospondin-1, sphingosine-1-phosphate and lysophosphatidic acid). Remodeling of the ECM with the help of a cross-linked fibrin/fibronectin/ vitronectin complex in damaged large vessels is observed both from their lumen and from the capillaries of the adventitial layer. This ECM remodeling creates rigidity in the vascular scaffold, which reduces its functionality.
Other key mediators that are important in vascular injury are angiotensin II, oxidized LDL (low density lipoprotein), proinflammatory mediators including IL (interleukin-1β, TNFα (tumor necrosis factor) and thrombin, circulating serine proteinases (plasminogen and plasma prekallikrein), which are activated to plasma plasmin and kallikrein, respectively [298,299]. Additional mediators are leukocyte proteinases such as neutrophil elastase, cathepsin G, matrix metalloproteinases (MMP -8 and MMP -9, mast cell proteinases, tryptase and chymase, and proteinases). vascular cells, including various MMPs (MMP-1, MMP -2, MMP- 3, MMP-12, MMP-13), Adamts-4 and Adamts-5 [276,300,301]. In the ECM, a class of MMPs are activated by serine proteinases derived as from plasma sources and from neutrophils, macrophages or mast cells.
Added to the overall picture of molecular transformations in the vascular wall is the periodic migration from the bone marrow and peripheral blood of a pool of precursor endothelial cells, monocytes and other white blood cells, which also produce growth factors. Blood islands of the bone marrow, consisting of hemangioblasts, are surrounded by endothelial cells. Primary capillaries are formed as a result of the fusion of blood islands to form primary capillary plexuses. Angioblasts are the main source of ECs with a strong ability to differentiate into functional ECs. Fusion of angioblasts determines vessel elongation. Endothelial cell precursors adhere to mature vascular endothelium and actively integrate into the vessel wall, differentiate into endothelial cells and, together with smooth muscle cells, form the vessel [56, 302].
Angiogenesis with the Combined Use of Growth Factors VEGF alone is known to inhibit pericyte coverage of vascular processes by inhibiting receptors on smooth muscle cells and led to destabilization of existing vessels [30]. The technology of loading multiple growth factors into a delivery system with different capacities and elution times is the preferred task for the researcher, taking into account the individual physical and physicochemical properties of each growth factor [303].
The combination of several growth factors has a beneficial effect on the morphology and stability of the engineered vasculature. Administration of angiogenic factors alone does not promote the formation of the pericyte layer, which is important for ensuring the mechanical stability of the vasculature [304].
According to the currently known points of application of regulatory peptides in angiogenesis and the development of tissue ischemia, a large role will belong to the use of factors with different mechanisms of action, including both interreceptor interactions and direct activation of signaling pathways in the cell through exogenous gene editing tools. The idea of using indirect control of multiple release of growth factors with good end results is known. For example, induction of hypoxia factor 1-alpha (HIF- 1a) stimulates the release of VEGF, bFGF and Ang-1, leading to improved blood vessel formation [305].
When using natural scaffolds that include two or more growth factors, it is important to consider not only the possible interactions between factors, factors and the matrix, but also the order of inclusion of proteins in the matrix, since opposite results can be expected [306,307]. It has been reliably established that polysaccharide scaffolds that contain three or more growth factors are able to sequentially release them into the ECM and more accurately mimic angiogenesis as in both in vitro and in vivo [17,306]. If it is known that VEGF and PDGF are powerful angiogenic factors with relatively different roles [56] and are involved in angiogenesis, respectively, in the early and late stages of vascular network formation, then when obtaining a transport system with these factors, it is necessary to take into account spatiotemporal profiles of their release. Factors that are responsible for stabilizing already formed early vessels must be included and activated at the late stage of angiogenesis [56]. If early PDGF activation occurs in the ECM when VEGF is highly expressed, then vascular endothelial growth factor may inhibit PDGF signaling where VEGFR-2 forms complexes with PDGFR-β, thereby interfering with its role in pericyte recruitment and new vessel stabilization [30].
When VEGF and FGF-2 are encapsulated in a hydrogel matrix containing PDGF microspheres, researchers obtain an early release of VEGF during the initial recruitment and proliferation of blood vessels and FGF-2 with different kinetic rates and delayed elution into PDGF medium. This sequence of growth factor release from matrices ensured the recruitment of mural and smooth muscle cells triggered by PDGF [307]. At the same time, analysis of the function of HUVEC cells indicates an accelerated transition from G0 to S phases of the cell cycle and improved distribution of cells in the G2/M and S phases. The synergistic effect of VEGF, FGF-2 and PDGF is confirmed by an increase in the migration distance of HUVEC compared to free scaffolds, ability to invade and form tubes [307]. The same pattern is observed in conditions in vivo using chicken chorioallantoic membrane. Delivery of VEGF, FGF-2, and PDGF from PDGF, VEGF, and FGF polysaccharide matrices resulted in a dramatic increase in the number of mature blood vessels in the thickened mesoderm. Cytokine analysis in the ECM with the participation of this cocktail of growth factors shows that the level of anti-inflammatory proteins in macrophages (IL -10) increases, and the expression of pro-inflammatory proteins (IL-12, NFB transcription factor) decreases or is completely blocked. This fact should be paid attention to, since the presence of a polysaccharide hydrogel in the ECM with the active participation of several angiogenic growth factors of endogenous or exogenous origin can locally suppress the severity of atherogenic inflammation against the background of hyperlipidemia [308].
The combined inclusion of growth factors in sulfated scaffold carriers and the creation of conditions for their dosed release into the medium leads to the formation of a high density of new capillaries [309-311]. This combination of growth factors is also justified due to not only the synergistic mechanism of action on the formation of the vascular network, but also the manifestation of various effects in angiogenesis aimed at one end result. For example, the inclusion of VEGF and FGF-2 in the transport system is aimed not only at the differentiation of endothelial cells ( VEGF), but also at increasing overall cell survival (FGF), despite the fact that both growth factors ensured microvascular density and in different degree of proliferation rate of endothelial cells and opsonization by pericytes [312-314]. The simultaneous inclusion of VEGF and ANG-1 in the transport system is aimed not only at inducing vascular permeability and destruction of the vascular membrane [315,316], but also at stabilizing blood vessels and stopping the leakage of blood plasma (ANG-1) [252]. Two growth factors ultimately restore blood flow in ischemic areas [317]. The combined use of PDGF and FGF -2 in the transport system also leads to a synergistic effect and to coordinated migration, proliferation of endothelial cells and neovascularization [318]. Moreover, different PDGF subtypes are able to coordinate chemotaxis and cell proliferation in a coordinated manner.
It is known that excessive expression of VEGF production by the vascular endothelium can form vascular angiomas, disrupting the normal course of angiogenesis. It was found that the co-expression of PDGF-BB and VEGF through a single viral vector does not affect the normal formation of capillaries and arterial vasculature not only when low and medium doses of VEGF are administered, but also specifically prevents the formation of aberrant vessels with the transition to angioma-like vascular structures at high doses VEGF [319]. This induced angiogenesis was maintained without any abnormalities up to 4 months of the experiment [320]. However, high levels of PDGF are characteristic of excessive cell proliferation, which may indicate the progression of atherogenic inflammation. The successful combination of VEGF and PDGF was demonstrated in, where dual release of VEGF and PDGF- BB was obtained with increased vascular density and maturity compared with the use of each GF alone. With this combination, high or low VEGF concentrations did not affect the final result; coexpression of both factors in each cell using a single vector resulted in revascularization, efficient growth of collateral arteries, increased blood flow, and decreased tissue damage [321]. This result indicates stable and normal angiogenesis [322].
Thus, creating a balance of stimulation between multiple growth factors results in coordinated function of multiple systems involved in active angiogenesis, for example, stimulation of vascular endothelial cells and recruitment of mural system pericytes. It has been established that the activation of pericytes is accompanied by the detachment and migration of these cells, which is observed during angiogenesis or tissue regeneration, when pericytes migrate to stabilize the developing vascular network.
Simultaneous delivery of VEGF, PDGF and FGF-2 via a polymer matrix increases in vitro angiogenic differentiation of human umbilical vein endothelial cells (HUVEC) and accelerates the formation of new vascular networks in the chorioallantoic membrane [322,323]. This consistent effect was confirmed by improved blood circulation in various animal models of ischemia of different localizations [324].
Integrins
Creation of signaling in a confined space at a local concentration of growth factors associated with many ECM proteins, immediately adjacent to the binding sites for cell adhesion, occurs through concerted stimulation of growth factor receptors and mechanoreceptors of integrins, which are typically colocalized in the membrane. Prior to the interaction of the integrin-growth factor system with the cell surface, many ECM proteins naturally bind both growth factors and integrins (αvβ3, αvβ5, α5β1) in a confined space due to adhesive structures [27, 325-327]. For example, the use of recombinant fragments of fibronectin is designed for the presence in one protein molecule of two binding domains for growth factor and integrin [328]. This tandem is designed to ensure co-localization and activation of integrins and GF s receptors in the cell membrane.
ECM protein-growth factor-integrin system can be included in a polymer carrier, which leads to a stronger synergistic effect in activating the cell signaling system. This phenomenon is explained by the ability of a polymer containing sulfhydryl groups in the molecule (for example, heparin) to reorganize the structure of the ECM protein [329,330]. This reorganization involves the unfolding of the protein with opsonized growth factor and integrin and making the integrin and GF binding regions highly accessible [331,332]. Such hydrogels have been found to provide local delivery and continuous release of sustained low doses of FGs in vivo. [333]. However, it has been established that in many cases the amount of growth factor in the vascular wall is extremely important, since initially high concentrations of the polypeptide can stimulate the formation of endothelial tubes with a stable capillary structure.
A wide variety of integrins, ligands, GFRs, and GFs themselves ensure the integration of signals between the two types of receptors. It has been established that integrins can have a potentiating effect when transmitting a signal not only through a specific receptor of the corresponding growth factor, for example, through VEGFR, but also through a number of other receptors, such as TGFβR, PDGFRβ, IGFR [334,335]. Moreover, one growth factor can activate several integrins. In addition, the mechanisms of independent activation of signaling pathways by both the growth factor receptor and one of the integrins are known. Independent activation may subsequently overlap and trigger the same signaling molecules. The interaction of several integrins with each other can activate the growth factor receptor without prior stimulation of the growth factor itself. Regulation of signaling pathways also includes processes of its attenuation, when cell enzyme systems are able to block phosphorylation of membrane growth factor receptors [336,337]. All these options for the formation of an intracellular signal require deciphering the mechanisms of transmembrane transmission of growth factors, especially with the participation of the kinase system in the area of focal adhesion, through a number of cytoskeletal proteins to the translocation of nuclear transcription. Thus, the diversity of interacting proteins in the ECM and transmembrane signaling means that the results of synergy or attenuation in signaling pathway formation must be considered as multilevel transduction [338,339].
Atherogenic Inflammation in The Vascular Wall and Beyond It
Lipid Metabolic Disorders
The most important vascular damaging factor is high levels of low- density lipoprotein (LDL), provided that hyperlipidemia occurs under sterile conditions without a bacterial component [340- 342]. LDL cholesterol level has remained the main determining factor in the occurrence and progression of atherosclerosis for many years [343,344]. Cholesterol is rich lipid pheroidal packets, coated with a phospholipid shell containing apolipoprotein B, transport water-insoluble cholesterol through the blood. The accumulation of LDL in the intimal space is provoked by a state of hyperlipidemia, which enhances the dysfunction of the vascular endothelium. Lipoproteins with a diameter less than 70 nm can cross the endothelial barrier and enter the arterial intima from the bloodstream [345].
However, in the initial stage of hyperlipidemia, when the endothelial cell monolayer is stable and at rest, specific integrins, due to their signaling ability, maintain the stability of the basement membrane in the matrix and suppress EC activation. At the same time, the stability and resting state of the endothelial lining of blood vessels is maintained by FGF (fibroblast growth factor)-2 and its main receptor FGFR1 [346,347].
Long-term hyperlipidemia causes endothelial cells to express cell adhesion molecules and endothelial growth factor and recruit white blood cells into the intima. Studies of cell transmigration across the endothelial layer suggest that neutrophils are among the first cells recruited to the inflammatory site, paving the way for subsequent engulfment of monocytes [348,349]. When analyzing the mechanisms of transmigration of white blood cells through the vascular endothelium, it is discovered that when animals receive a cholesterol diet under conditions of hyperlipidemia, the number of neutrophils rolling along the endothelial layer with the surface expression of sticky clusters increases. At the same time, the speed of rolling monocytes decreases [350]. This transformation of the leukocyte membrane increases their contact with the endothelium [351].
After the leukocytes have coagulated and are firmly attached to the endothelium, the process of transmigration begins. Leukocyte translation is achieved through stable interactions between leukocyte integrins and endothelial adhesion molecules [352]. It is assumed that for complete adhesion of leukocytes to endothelial cells, a temporary interaction of chemokines and glycosaminoglycans is formed to retain chemokines in the glycocalyx space near the endothelium. Strong adhesion of leukocytes and vascular endothelium is carried out due to the interaction of integrin with the molecules of intercellular ICAM-1, ICAM-2 and ICAM-3 and vascular intracellular adhesion VCAM- 1 with the participation of lymphocyte-associated antigen (LFA- 1) and macrophage receptor (Mac-1) [353,354]. The retention of leukocytes on the surface of the endothelium is the beginning of transmigration into the subendothelial zone. Such transmigration is accompanied by the formation of clu3sters consisting of ICAM- 1 and VCAM-1 molecules [355,356]. Adhesive clustering changes the size of EC intercellular contacts, reduces the size of ECs themselves with the help of structural proteins of the cytoplasm, creates conditions for paracellular transport of leukocytes, actively compressing the nuclei of leukocytes with the help of endothelial actin filaments [357]. This conformation of the endothelium forms endothelial protrusions around the infiltrating leukocyte [358]. In this case, ECs reduce the expression of adhesive cadherin [359,360].
Transmembrane migration of leukocytes through thin transcellular pores of the EC cytoplasm, rich in caveolin 1 and adhesion molecules ICAM-1, is known [359,361]. In this case, clustering of adhesion molecules on EC is accompanied by the release of pro- inflammatory cytokines from leukocytes: IL-1β, IL-6, C-C motif chemokine ligand (CCL 2) and C-X-C motif chemokine ligand (CXCL 8) [362].
Monocyte Transmobilization
Monocytes of the vascular bed are translated into the subendothelial zone and greatly increase the uptake of already oxidized lipid mass through scavenger receptors or phagocytosis of aggregated lipoproteins, turning into residual macrophages [363,364]. The oxidized fraction of LDL is delivered to lysosomes for breakdown into cholesterol and fatty acids [365]. Lysosomal hydrolysis of cholesteryl ester results in the formation of unesterified cholesterol molecules, which are then released from the lysosomes and re-esterified in the cytoplasm, followed by the formation of cholesteryl ester droplets in the cytoplasm, leading to the formation of foam cells. Cellular excess of unesterified cholesterol is toxic to macrophages, so storage of cholesterol in the form of cholesteryl ester droplets is considered a protective response [366].
It has been shown that the translation (transmigration) of monocytes from the bloodstream through the endothelial lining is selectively regulated by E-selectin [367]. The intracellular actomyosin machinery forms integrin-rich protrusions to target monocyte chemotaxis through the endothelial layer, scanning the endothelial lumen at sites of extravasation [368].
Impaired production of reactive oxygen species and reactive nitrogen species causes the loss of LDL's ability to interact with LDL receptors. This significantly increases the affinity for scavenger receptors such as CD36 and lectin-like oxidized LDL receptor-1 (LOX-1), SR-B1 on ECs, vascular smooth muscle cells and macrophages [369-371]. It was found that the expression of high-performance scavenger receptors and their high affinity for LDL particles do not decrease with increasing cholesterol content in the cell. This allows macrophages to be overloaded with cholesterol esters and enhance the formation of foam cells, as signs of early atherosclerotic damage. Excessive accumulation of cholesterol can lead to the formation of cholesterol crystals, inducing the breakdown and secretion of IL-1β and IL-18 [372]. Numerous studies support the concept of the ability of oxidized LDL particles (oxLDL) to form atherogenesis [373]. Studies show that oxLDL suppress the activity of vascular endothelial cadherin (VE-cadherin) and upregulate platelet-endothelial complex adhesion molecule (PECAM-1), which promotes monocyte transmigration [374].
It has been established that two growth factors are required for the differentiation of monocytes into macrophages: macrophage colony-stimulating factor (M-CSF) and granulocyte-macrophage colony-stimulating factor [375,376]. The accumulation of lipids in macrophages transforms their cytoplasm into a foamy substrate. The cell mass, along with the endothelial layer, becomes a source of proteolysis and, before degradation, is enclosed in a mass of interstitial collagen matrix with the formation of a fibrous cap. This newly formed construct in the intimate zone near the endothelial layer contains denatured protein substrates, FNs, osteopontin, tenascin C and a number of proteoglycans bound by macrophages through the expression of integrins αMβ2, axβ2, α4β1 [377]. The concentration of intimal proteoglycans, in addition to residual macrophages, determines the localization and accumulation of LDL in areas of intimal thickening [378]. The accumulation of a mass of oxidized forms of LDL in the subendothelial space maintains high adhesiveness of endothelial cells and increased lipid and cellular leaks [378]. Cellular and lipoprotein leaks occur in intimal areas under shear stress. In these areas, turbulent blood flow changes the cellular arrangement of ECs. Tight interendothelial junctions become “leaky” and their permeability to large molecules increases. The dehiscence of intercellular junctions promotes the uptake of plasma lipoproteins rich in LDL and triglycerides (TG) either transendothelially or by diffusion at intercellular junctions [379].
Modified lipids activate inflammatory cells in the intima, which produce tumor necrosis factor (TNF-alpha), interleukin-1, -4 and -6 and interferon-gamma, which activate both endothelial cells with endothelial surface expression of adhesion molecules (VCAM, ICAM, E-selectin) and smooth muscle cells of the mural zone in the vessel wall. The accumulation of LDL cholesterol by marophages is accompanied by a self-sustaining inflammatory mechanism, since ox LDL irritate endothelial cells, increasing the production of adhesion molecules. High levels of reactive oxygen species (ROS) and reactive nitrogen species (RNS) convert LDL into ox LDL, which is embedded in the intimal layer of the vascular wall. Thus, the established regulation of the relationship between foamy and non-foamy macrophages in an atherosclerotic plaque ensures a constant flow of monocytes and LDL into the damaged area, which means plaque growth [380]. Therefore, high levels of oxLDL in the arterial vessel wall can be regarded as a clinical marker of plaque inflammation [381,382]. Macrophage proliferation in response to macrophage colony-stimulating factor (M-CSF) is accompanied by macrophage engulfment of foam cells. This process means secondary cell necrosis and inflammation. In addition, the proliferation of macrophages in lesions may be due to the production of M-CSF secreted by smooth muscle cells. In addition to the direct effect of macrophage oxLDL on endothelial cells, the cells produce the vessel-specific inflammatory enzyme phospholipid-associated phospholipase A2 (Lp-PLA2). The enzyme is produced in atherosclerotic plaques, circulates as part of apolipoprotein B, hydrolyzes oxidized phospholipids on LDL particles in the intima of the artery, causing the formation of mediators of pro-inflammatory and pro-atherogenic properties. Thus, the cellular composition of an atherogenic plaque self- sustains the production of oxLDL, a high content of hydrolytic enzymes and the production of inflammatory mediators. This process creates long-term endothelial dysfunction and progression of atherosclerosis [383].
Cellular invasion involving the full thickness of the arterial vessel involves infiltration of VSMC into the intimal space. The accumulation of LDL particles in the subendothelial space and the formation of aggregates upon binding of particles to proteoglycans provokes entry into smooth muscle cells through the LDL receptor superfamily of related proteins (LRP). Attention should be paid to the possibility of uptake of LDL and glycosaminoglycans, which contribute to the accumulation of lipids in the intima. Thus, in addition to macrophages, smooth muscle cells can also accumulate cholesterol, forming the LRP superfamily, while retaining the ability to evade expression inhibition mechanisms under high cholesterol conditions. It has been demonstrated that smooth muscle cell metaplasia can also give rise to foam cells resembling macrophages [384]. Their localization is typical in the human intima, especially in areas of atheroma formation. In this regard, the saturation of macrophages and smooth muscle cells with cholesterol in the mural zone of the vessel leads to the progression of the lesion [385]. Cellular invasion into the layers of the vascular wall occurs due to the interaction of almost all serine proteinases with FN, vitronectin, fibrinogen, fibrin, provokes and supports proteolysis, activation of the MMPs cascade and the release of heparin from adventitia mast cells into the ECM . Both factors stimulate the release of active growth factors. Activation of the MMPs cascade causes regression of the EC of the vascular tube [386] (Figure-scheme 1, Figure-photo 2A, 3BC, 4DE, 5F, 6G, 7H).
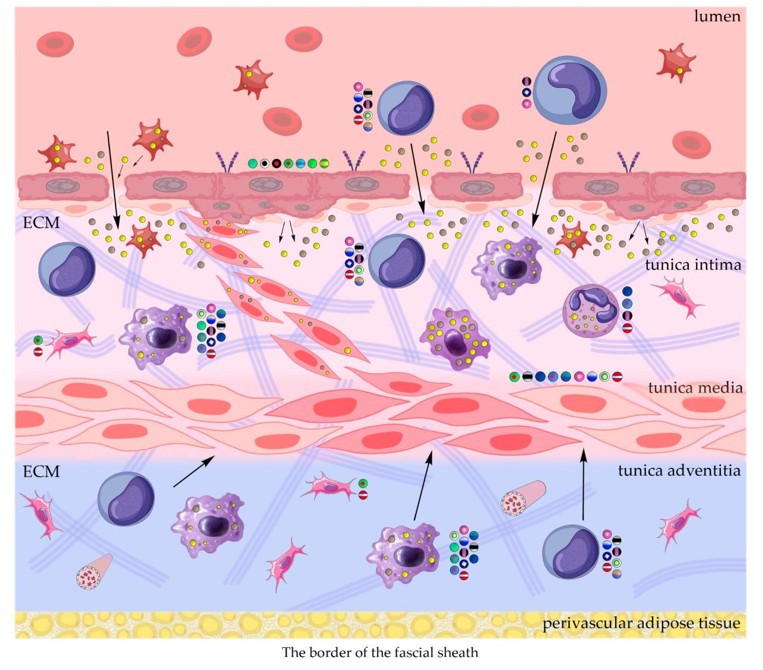
Figure-Scheme 1: Hypothesis. The level of activity of cellular growth factors in the vascular wall during atherogenic inflammation. During atherogenesis - massive dense adhesion of leukocytes to the vascular endothelium with the help of clusters: integrin – ICAM-1, VCAM-1. A change in the amount of intercellular contact of the vascular endothelium as a result of reduction of the cytoskeleton of cytoplasmic proteins and compression of leukocyte nuclei is the effect of interendothelial transport of leukocytes. Subsequent penetration of masses of mnocytes and low-density lipid spheroids containing plasma-insoluble cholesterol. Active capture of cholesterol crystals from LDL by monocytes-macrophages, including oxidized forms, suppression of cholesterol reverse with a high content of oxLDL in cells, formation of “foam” cells. Proliferation and migration of smooth muscle cells into the intimate zone with signs of cholesterolization of the cytoplasm, thickening of the intima. Weak cellular reaction of the adventitial and sub-adventitial layers. Despite the activation of angiogenesis when determined by pro-atherogenic growth factors, there is an active filling of the intimate zone with inflammatory infiltrate with weak signs of migration of type M1 macrophages filled with LDL and cholesterol crystals into the adventitia zone and beyond its borders. Active cell adhesion of the endothelium and the angiogenic reaction in atherosclerosis is accompanied by massive infiltration of leukocytes and monocytes through the vascular wall. The process of recruitment of cells in the mural zone is low; there is no mass formation of microcapillaries with lumen and blood flow.
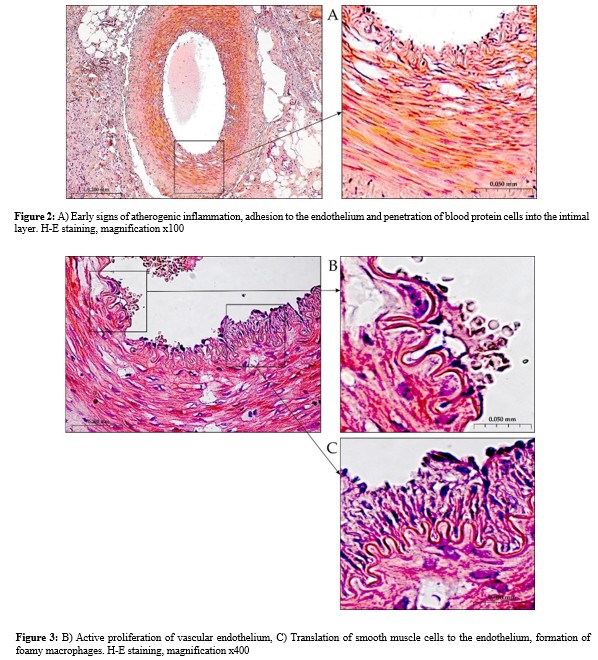
Figure 3: B) Active proliferation of vascular endothelium, C) Translation of smooth muscle cells to the endothelium, formation of foamy macrophages. H-E staining, magnification x400
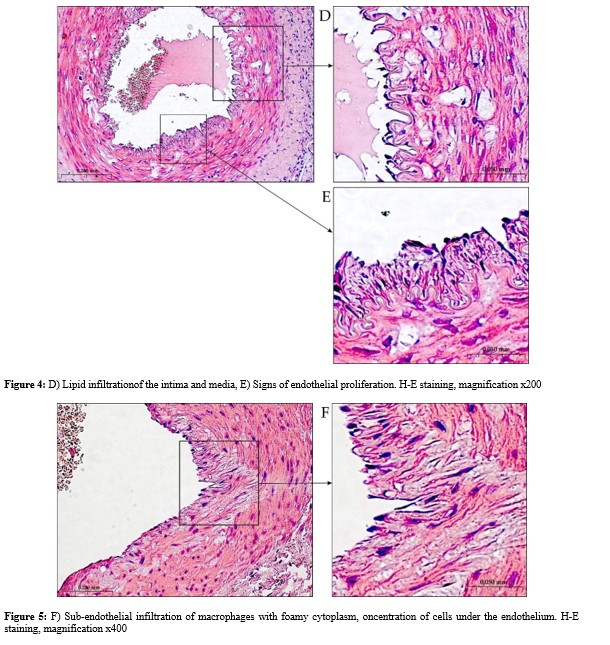
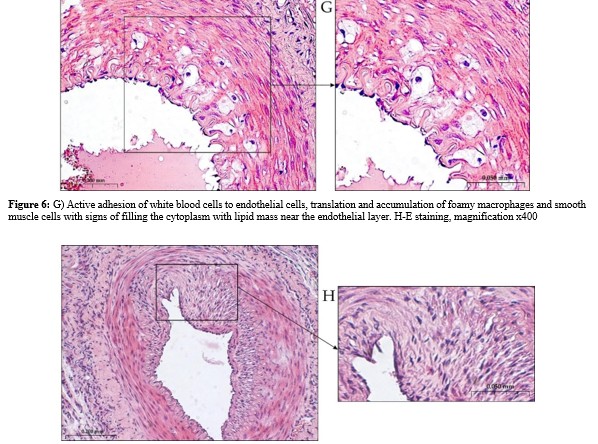
Figure 7: H) Atherogenic damage to the arterial wall in a rabbit fed a cholesterol diet (ChD) for 4 months [387]. Coloring H-E. The thin histological sections (3–4μm) using a Leica automated system (Germany). Microphotographs were obtained using an Axio Imager A1 microscope with an AxioCam MRc5 photography system and an Axio Vision software (Carl Zeiss, Germany). Cross section of the wall of a rabbit artery is affected by atherogenic inflammation, thickening of the intima, migration and proliferation of endothelial cells, formation of “foamy” phages, migration of smooth muscle cells to the vascular endothelium. Accumulation of xanthoma and smooth muscle cells under the vascular endothelium, formation of soft atherogenic plaques. Translation of smooth muscle cells towards the vascular endothelium, weak cellular reaction in the adventitia and beyond. H-E staining, magnification ÃÃÃÃÂ?????200.
In advanced lesions of the vascular wall, foam cells easily degrade and release their contents into the extracellular space, thereby worsening the inflammatory status and structural stability of the plaque [388]. Intense proteolysis thins the fibrous cap and provokes rupture of the endothelial layer. In this case, attention should be paid to the fact that when analyzing the dislocation of VSMC in the intimal space, the absence of these cells at the site of the future rupture of the atherogenic plaque is revealed. [389,390]. In connection with the results of these studies, it becomes important to determine the levels of specific MMPs during the formation of the initial stage of an atherogenic plaque in the wall of a large arterial vessel in order to develop new technologies for local regression of atherogenic inflammation.
T-cells
In human atherosclerotic plaques, T-cells occupy a significant part. Among the subpopulations of T-cells, various subsets of T-helper cells play an important role [391]. The growth of atherosclerotic plaques is associated with the participation of Th [392-394]. Together with memory T-cells, Th make up the majority of T-cells found in atherosclerotic plaques [395]. The population of T-regulatory cells (Tregs) is capable of expressing the transcription factor forkhead box protein P3 (FOXP3) and CD25, which exhibit strong anti-inflammatory properties, suppress the proliferation of Th cells, and induce an anti-inflammatory phenotype of macrophages [396,397]. Expression of IL -10 and transforming growth factor (TGF)-β by Treg cells exhibits antiatherogenic effects in atherosclerosis [398]. The process of migration into the vascular wall containing atherogenic plaques, circulating T-cells enter with the participation of endothelial P-selectin, using the glycoprotein ligand of platelet selectin (PSGL-1) [399], with the support of cofactors CD43 and CD44 [399-401]. Thus, a number of chemokines with their receptors with dislocation in intraendothelial vesicles attract T-cells, forming tight junctions with endothelial cells, and are translated into the subendothelial zone [402]. It is possible that the translation of these cells is similar to the transmigration of other leukocytes, accompanied by the expression of protein adhesion molecules.
B cells
B-cells are also involved in local immune responses that contribute to chronic inflammation in atherosclerosis [403]. However, research confirms that B cell subsets may exhibit both antiatherogenic and proatherogenic properties.
Platelets
Activation and aggregation of platelets in affected vessels is also subject to shear stress, especially at the bifurcation points of the great arteries. Platelet adhesion to the endothelium is induced by the expression of adhesion molecules already in the early stages of atherosclerotic lesions [404]. Reduced efflux of LDL cholesterol from platelets leads to their activation. The adhesiveness of these cells to the vascular endothelium is directly stimulated by the oxLDL fraction through binding to CD36 [405]. At the same time, hyperlipidemia increases platelet aggregation activity [406]. Platelet activation increases the expression of P-selectin and CD40 ligand, which increases adhesiveness to the vascular endothelium and monocytes at problem points. In this case, the monocyte- platelet complex maintains strong contact and penetrates the intimate area and participates in the formation of the core of the atherogenic plaque. This mechanism appears already at the early stages of atherogenesis [407,408].â??â??â??â??â??â??â??
Macrophages
Two main subtypes of MΦ are widely recognized: proinflammatory macrophages (M1) and anti-inflammatory macrophages (M2). A microenvironment containing the cytokines interleukin 4 (IL-4), interleukin 13 (IL-13), interleukin 10 (IL-10), IL-1RA, PDGFC, TGF-β, FGL2 promotes M2 polarization. Contact with a platelet induces a pro-inflammatory profile of the macrophage, triggering membrane surface expression of IL-1β, IL-6, IL-12 interferon-γ (INF-γ), tumor necrosis factor (TNF-α) [409]. Lipopolysaccharides also influence the development of the M1 phenotype [410]. Pro-inflammatory macrophage phenotype increases the area of cellular necrosis in the atherosclerotic core in an experimental model mice. Massive penetration of the platelet-leukocyte complex through the vascular endothelium is mediated by the expression of adhesion molecules on the surface of endothelial cells and the emergence of a signaling pathway based on platelet-endothelial cell adhesion molecule (PECAM-1) [411]. The mechanism of activation and translation of the platelet-monocyte complex is similar to the transmigration of platelet-lymphocyte aggregates. Delivery occurs through platelet microvesicles to monocytes and neutrophils [412].
Along with the role of scavenger receptors on leukocytes, stimulating the uptake of oxLDL by macrophages, platelets are also capable of synthesizing the motivation chemokine lectin C-X-C (CXCL4), which is used to directly take up oxLDL by macrophages [413]. In addition, it has been established that platelets, in addition to producing direct angiogenic factors, synthesize a number of lectin mediators that regulate intercellular interactions during the development of atherogenic lesions in the vascular wall.
Dendritic Cells
Dendritic cells (DCs) are a subtype of antigen-presenting cells (APCs) that are found in the intimal and adventitial spaces of arteries [414]. DCs are perceived as the central link of adaptive immunity in atherosclerosis. These cells capable of monitoring antigens directly adjacent to the endothelial lining [415]. These cells circulate in human peripheral blood and are morphologically similar to rodent dendritic cells. DC subtypes are generated in the bone marrow from pluripotent hematopoietic stem cells and share a common progenitor [416]. From the bone marrow, DCs migrate to peripheral lymphoid and non-lymphoid tissues [417] due to their own expression of chemokine receptors and ligands [418]. It has been observed that DCs in the vasculature accumulate in areas of atherogenic plaque formation, at points of turbulent blood flow, in the marginal parts of the nucleus, and with smaller amounts in the central part of the unstable plaque [419-421].
Like macrophages, DCs are capable of accumulating intracellular lipids and transforming into foam cells upon experimental activation of the hypoxic inducing factor HIF-1α. Depending on the level of hypoxia, as well as the levels of stimulation of proatherogenic and antiatherogenic cytokines, stimulation of proatherogenic or antiatherogenic T-cell lines occurs [422,423].
Angiogenic, Proatherogenic And Antiatherogenic Growth Factors in Atherosclerotic Lesions of the Vascular Wall Research confirms that the profile of fluctuations in the activity of proinflammatory cytokines in the blood plasma corresponds to the level of cytokines in the intimate zone and in particular in the atherogenic plaque [424].
Growth factors are secreted by cells and can stimulate their functions through autocrine or paracrine mechanisms. These structures are short-distance polypeptides and act locally [425]. The high affinity of GFs for many proteins in the tissue compartment ensures slow diffusion, cell attachment and signaling into the cytoplasm and nucleus of the cell [426]. This affinity of matrix extracellular proteins is due to the presence of binding sites for growth factors. Such stationary proteins include type IV collagen, fibronectin, and vitronectin [427-429,431]. A number of adhesive proteins, such as laminin, tenascin, thrombospondin, fibrillin, play the role of secondary ligands that are directly recognized by growth factor receptors and induce transmembrane signaling. Such adhesion molecules form connections between cells and surrounding scaffolds, serving as external transduction of specific signaling [432]. In turn, cells in contact with exogenous material also express a number of adhesive structures, such as integrins, immunoglobulins, cadherins, and selectins, on their surface and in the ECM [433].
Thus, the ECM is central to regulating the availability and activity of GFs in vivo, in which GFs are found tightly bound to the ECM [434] and are released through proteolytic degradation near matrix adhesions. In essence, the ECM can be considered as a depot of GFs as a result of sequestration of molecules, a zone of regulation of local concentration and a zone of stable protection of their original activity. Otherwise, the introduction of a water- soluble form of the growth factor without prior connection with the delivery system abolished the effect of complex presentation. Thus, communication between cells in the choroid is mediated through growth factors and their cellular receptors [435,436]. In this regard, precise control of the signaling of these factors in the local area could potentially allow control of the regenerative process. Based on this assumption, the determination of the levels of GFs in the vascular wall will also indicate the processes of stimulation or inhibition of atherogenesis and angiogenesis. If we take into account such a factor as VEGF, it is important to note that inflammatory processes in blood vessels (diabetes mellitus, chronic long-healing wounds, ulcers) are accompanied by a very low level of growth factor. Therefore, maintaining local VEGF concentrations is critical for angiogenic efficacy. The authors observed a consistent restoration of the vasculature within 2 weeks after VEGF delivery in an experiment on mice [246]. It has been established that the ECM and the recruitment of mural cells are important for the stabilization of nascent endothelial tubes and capillaries. In order to preliminarily know the level of VEGF in the ECM, it is sufficient to determine the levels of placental growth factor, fibroblast growth factor-2 and hepatocyte growth factor. It is known that the induction of these factors in the system in vitro indicates an increase in VEGF levels [437].
Under physiological conditions, the arterial endothelium is covered with a glycocalyx, a layer of proteoglycans and extracellular matrix components, which has properties that inhibit platelet activation, prevent thrombus formation, and promote thrombolysis [438,439]. Thrombomodulin and heparan sulfate proteoglycans on the endothelial surface and the production of nitric oxide and prostacyclin by endothelial cells provide the anticoagulant and antithrombotic properties of the normal endothelial monolayer. Ischemia of any degree means the secretion by endothelial cells of not only direct angiogenic growth factors, but also the release of cytokines such as monocyte chemoattractant protein 1 (MCP- 1), cell adhesion molecules (ICAM-1 and VCAM-1), granulocyte- macrophage colony-stimulating factor (GM-CSF).
Despite the temporary thickening of interendothelial contacts in the zone of increased lateral branching of vessels above the site of occlusion, local critical ischemia develops in the periphery below the zone of obstruction to blood flow [440]. With atherosclerosis and the formation of soft and stable plaques, these mechanisms for compensating the blood supply to the lower limb are disrupted, which is subsequently indicated by the lack of formation and stabilization of newly formed vessels. Violations come primarily from the state of tissue hypoxia. Lack of oxygen as a substrate blocks hydroxylation by the prolyl hydroxylase domain (PHD) family and degradation of the hypoxia-inducible factor 1 (HIF- 1α) subunit. Under normoxic conditions, HIF-1α is inactive due to both ubiquitin-mediated degradation and transcriptional inhibition.
During hypoxia, as a result of suppression of prolyl hydroxylase activity, HIF -1 α accumulates and activates, forming a heterodimer with the HIF-1β subunit. The HIF-1 molecule stimulates the transcription of angiogenic factors (vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF) and stroma- derived factor-1 alpha (SDF-1α) in endothelial cells as targets [441,442]. As targets for HIF -1s that are involved in angiogenesis are considered VEGF-A, PDGF-BB, stromal cell factor-1 (SDF- 1) and endothelial nitric oxide synthase eNOS), as well as heme oxygenase-1 (HO -1) [443-447]. As a result, the activity of specific and nonspecific angiogenesis factors increases [443-445].
An imbalance in the expression of pro-atherogenic (pro- inflammatory) and anti-atherogenic (anti-inflammatory) cell markers while maintaining hyperlipidemia conditions stabilizes destruction in the subendothelial space. Expression of receptors for adhesive factors of residual blood cells under the vascular endothelium and the vascular endothelium itself during the progression of ischemia: endothelin-1, caveolins-1,2,3, selectins P (CD62P), E (CD62E), L (CD62L) and antibodies to them on vascular endothelium, lymphocytes and platelets, intercellular adhesion marker (ICAM-1), vascular adhesion marker (VCAM- 1), monocyte chemotaxis protein (MCP -1), monocyte colony stimulating factor (MCSF), pleiotropic cytokine (TNF-α), C-reactive protein (CRP), platelet-derived growth factor (PDGF), interleukin (IL) family in atherosclerosis create a picture of multicellular destruction affecting all layers of the main vessel, including the adventitial layer [220,448-496]. An important comparative analysis showed that the level of proinflammatory cytokines circulating in the blood plasma correlates with the level of cytokines in the atherogenic plaque [497]. At the same time, multifunctional interferon that activates macrophages (IF-gamma), as well as IL-6, IL-12 and IL-15, tumor necrosis factor (TNF-a) are highly expressed by NK cells and T-cells in the soft plaque compared to unaffected artery, exceeding the levels of activity of anti-inflammatory cytokines such as IL-4, IL-10 [424,498,499]. Among cytokines, IF-gamma is the most important factor in the pathogenesis of atherosclerosis, since this marker plays a multifunctional role in the development of both early (VCAM-1, ICAM-1 factors, cytokines, chemokines, class I and II expression inducing antigens of the major histocompatibility complex (MHCs) on macrophages, T-lymphocytes, NK cells), and in the late stages of atherogenic inflammation, including the formation of a pool of macrophages saturated with oxidized fractions of low- density lipoproteins (oxLDL), accumulation of cholesterol in an atheromatous plaque with activation of cytokines, NO synthase of monocyte chemoattractant protein (MCP-1), increasing the synthesis of proteins of the class of metalloproteinases (MMPs) [500,501]. Polarization of primary macrophages by interferon gamma increases the expression level of the inflammatory cytokine interleukin-1β (IL-1β), thereby inducing angiogenesis by activating VEGF-A transcription in HLI mouse models. Deficiency of the gene that controls the synthesis of INF-gamma during experimental atherogenesis leads to a reduction in pathomorphological disorders in the vessel wall [502]. Massive uptake of oxidized forms of LDL by macrophages is ensured mainly by the expression of scavenger receptors class A (SR-A) and B (SR-B1, CD 36) [503-505]. Any artificial external intervention in this pathological molecular- cellular cascade for the purpose of therapy is designed to break the interconnected processes of progression of atherogenic inflammation. As a result of this postulate, the project's scientific team sets as its main task the determination of the balance between the levels of pro-atherogenic and anti-atherogenic molecules in the vascular wall in animals receiving and not receiving a cholesterol diet, as well as in animals with implantation of a polysaccharide sulfated polymer into the vascular lesion area.
These natural mechanisms for compensating for ischemia in case of serious circulatory disorders cannot provide a high level of angiogenesis [38,506]. Despite promising angiogenic results using growth factors such as VEGF in experimental settings, controlled clinical trials using various carriers and growth factors using standard technology to deliver glycoprotein to ischemic tissues in patients have not shown the prolonged expected benefits [507,508]. angiogenesis during intravenous or intracoronary infusion of the drug [509,510]. Administration of VEGF at a safe dose did not induce sufficient angiogenesis to correct cardiac or skeletal muscle ischemia [511]. The same neutral result was obtained using FGF 165 and FGF 2 [512,513].
Cell Functions in A Soft Atherogenic Plaque with Hypercholesterolemia in The Intimate and Middle Zones of The Vascular Wall
It is argued that atherosclerotic lesions are characterized by the lifelong accumulation and transformation of lipids, inflammatory cells, smooth muscle cells and necrotic cell debris in the intimal space under a monolayer of endothelial cells [514]. However, researchers continue to try to find effective mechanisms for controlling the functions of the ECM and creating a reversal of atherosclerotic lesions of the vascular wall. It is known that the process of cholesterolization of the intimate zone of the vascular wall is accompanied by the proliferation of smooth muscle cells in the middle layer of the vessel with a violation of their orientation and migration to the intimal zone. The expression of residual blood cell receptors and multicellular destruction in all layers of the vessel indicates that the inflammatory reaction extends beyond the adventitia [424,497]. The layer of the vessel adventitia is functionally associated with the middle and intimal layers. However, engineering of the vascular adventitia is able to regulate the proliferation of smooth muscle cells in the middle and endothelial layer of the vessel, which is important in the management of atherogenic inflammation [515,516]. Targeted implantation of cholesterol-affinity polysaccharide constructs into the paravasal sheaths of the main arteries over a large area of the vessel will likely be aimed at exogenous regulation of cell functions directly related to the formation of soft plaques [517]. In this case, the drainage capabilities of the adventitial lymphatic capillary bed can play an important role, and therefore, perform atheroprotective functions by removing lipids contained in plaques during regression [518].
It is known that the progression of plaques is accompanied by the accumulation of inflammatory cells in the adventitial layer of the vessel wall. As a result of this cellular infiltration, the adventitial vasculature expands as atherosclerosis progresses [290,519]. Experimental disruption of drainage by lymph nodes and vessels of the interstitial fluid of the mural zone of the intimate space leads to a concentration of CD 3 + T-cells not only in atherogenic plaques, but also in the adventitia itself, increases the thickness of the intimal layer, which leads to the progression of atherosclerosis. Moreover, lymphatic dysfunction precedes the onset of atherosclerosis [520]. It has been established that the greater the degree of damage to the vascular wall by the atherogenic process, the higher the concentration of lymphatic capillaries and nodes in the adventitia. In connection with this result, analysis of the number and characteristics of lymphatic vessels in the adventitia is an important marker of the activity of atherogenic inflammation and corresponds to the degree of damage to the intimal membrane [521-523].
Circulating Monocytes
Circulating myeloid cells actively interfere with the processes of induction and maturation of new vessels [524]. VEGF expression recruits monocytes, which are retained in the perivascular zone through stromal cell factor-1 (SDF-1) signaling and secrete proangiogenic factors [525]. Circulating monocytes induce invaginal vascular growth, form and stabilize intraluminal columns, and increase the vasculature [526]. The movement of monocytes between pericyte filopodia completes recruiting angiogenesis by fusion and anastomosis of tip cells [21]. Transformation of circulating monocytes into stationary macrophages in the ECM retains the ability to express the proangiogenic receptors Tie 2 and Neuropilin1 (Nrp1) and recruit smooth muscle cells. Expression of Nrp1 induces interaction with VEGF and thus attracts smooth muscle cells to the area of VEGF expression. Completion of arteriogenesis occurs with the participation of Ang1, TGF-β and PBGF-BB [527].
Smooth Muscle Cells (VSMCS) And Residual Macrophages The main events in chronic ischemia involve the interaction of polypeptide growth factor molecules in the ECM and endothelial cells, as well as the middle layer of the vascular wall. Short and slow local diffusion of growth factors through the extracellular matrix implements specific receptor intercellular and transcellular signals for cell differentiation, proliferation and migration [425]. This process involves the layer of smooth muscle cells (VSMCS). If moderate autophagy of VSMCS and mild endothelial reticulum (ER) stress support compensation of angiogenesis, then with the progression of tissue ischemia in the arterial wall, morphological signs of proliferation and migration of VSMCS from the midzone of the vessel to the intima and vascular endothelium appear [528]. It is known that these cells can undergo transdifferentiation into macrophage-like cells with the uptake of lipids and transformation into “foam” cells [529-531]. Moreover, VSMCS can take up cholesterol from the environment of macrophage foam cells using membrane-derived particles [532].
Detailed studies of the interaction between marophages and VSMC during co-culture of cells under in vitro conditions vitro show that supernatants contain increased levels of TNF-α and IL-6, IL-1β, MMP-2, MMP-9 obtained from both macrophages and VSMCs [533]. In this case, VSMC induced the production of reactive oxygen species (ROS) [534]. If macrophages saturated with lipoproteins and VSMCs are cultured in the same container, then after 5 days one can observe the saturation of VSMCs with lipids as a result of intercellular lipid transfer [535]. The same pattern can be observed when culturing LDL -replete macrophages with VSMCs [536].
As atherosclerosis progresses, VSMC proliferation and secretion transform the plaque. The composition of the plaque largely consists of VSMC and their synthesis products (elastin, collagens, proteoglycans. In addition, the composition includes cellular elements and metabolites of the inflammatory reaction [537].
The influx of active phagocytic cells into the intimate zone of the artery through interendothelial contacts and transformation into macrophages signifies the stage of vessel remodeling, the active uptake of cholesterol and low-density lipoproteins [38]. This process in the early stages of soft plaque formation is accompanied by macrophage opsonization with chemoattractants and high autophagy activity. Active angiogenesis in the area of a soft atherosclerotic plaque forms many microvessels with an influx of a pool of monocytes and macrophages. However, the process is eventually replaced by a picture of threshold ER stress, a reduction in cytoskeletal proteins of neighboring smooth muscle cells, a decrease in autophagy and angiogenesis, a change in the synthesis profile to collagenogenesis and the secretion of products into the extracellular space [538]. Excessive proliferation of VSMCS in the intimal zone is accompanied by an increase in apoptosis and the release of cellular detritus products into the interstitium. It should be said that the formation of a fibrous dome of a soft atherogenic plaque is complemented by the formation of a lipid core under the dome as part of a monocyte-macrophage cell mass. An active influx of leukocyte mass through interendothelial leaks and active transport of opsonized leukocytes with chemoattractant and cell adhesion molecules fill the atherogenic core. Hyperlipidemia, combined with high production of ROS, glycosylates and oxidizes lipoproteins, ensuring the transfer of cholesterol and low-density lipoproteins to the intimal zone with the participation of chemoattractants. As stated above, leukocyte cells express scavenger receptors that are capable of capturing molecules of oxidized lipoproteins that have penetrated the intima. In the intimal membrane, active utilization of oxidized lipoproteins occurs by the macrophage mass and the formation of cells with “foamy” cytoplasm. It is important to note that at the early stage of the formation of atherogenic inflammation, active intervention in the process of angiogenesis can restore blood perfusion. Such evidence has recently been presented in a number of papers [539- 541].
However, the role and impact of angiogenesis in atherosclerosis remains an unresolved problem. The reverse development of atherogenic inflammation and the formation of a healthy neointima in researchers is associated with neovascularization. The formation of new microvessels under the fibrous dome of a soft atherogenic plaque is a natural response to increasing ischemia. At the same time, there is a version that neovascularization is one of the effective factors in the growth and rupture of atherosclerotic vessel lesions [289,542]. Nevertheless, there is still no convincing evidence that the angiogenic response causes the formation of atherosclerotic plaques, their growth and progression to the stages of atherosclerosis [542]. At the same time, as the active formation of microvessels with the support of molecular promoters (CXC-1, -2, -3, -5, -6, -7, -8; FGF-1, -2; HGF; HIF -1, -2,- 3; PDGF-A, - B,-C; TGFβ1, -2, -3; VEGF-A, -B, -C, -D, PLGF, and stabilization of the finished vascular network using angiogenesis inhibitors (CXC-4 , -9, -10, -12, -14; Angiostatin; epithelium - derived factor (PEDF); Thrombospondin (TSP-1, -2, -3, -4, -5) (COM) cannot clearly indicate the progression of atherosclerotic inflammation with plaque formation and growth [543].
Mechanisms of Regulation of Lipid Metabolism in The Vascular Wall in Case of Using Natural Polysaccharides. The Role of Growth Factors in The Regulation of Atherogenic Inflammation
The atherogenic inflammatory reaction is characterized by the deposition of a free mass of cholesterol crystals in the intimate zone of the vessel. At the same time, cholesterol accumulation occurs in the cytoplasm of the monocyte-macrophage mass, which has penetrated into the interstitial zone from the systemic circulation, due to hyperlipidemia and high cell adhesion to the vascular endothelium.
The question on the agenda of modern research is: how does the accumulation of lipids in the subendothelial space contribute to inflammation? It is necessary to point out that the general biological law of nature about the active translation of biological fluids (blood, plasma, interstitial fluid, cerebrospinal fluid, intestinal chyme, urine, bile) indicates the absence of an inflammatory reaction in the absence of an obstacle to movement, as well as in the case of active drainage. Blocking of translation for various reasons (mechanical or dynamic intestinal obstruction, blocking the outflow of milk secretions, congestion in the liver, impaired myocardial contractile function, impaired passage of secondary urine or impaired drainage function of macrophages in the ECM) is always a harbinger of inflammation. Based on this postulate, changing the translation pathway of “foam” cells and cholesterol in the vessel wall can be achieved by artificially placing nano-sized polysaccharide ingredients in the perivascular space and forming a long-term drainage of inflammatory reaction metabolites. In the area of the so-called secondary ECM, not only drainage of interstitial fluid can be created using the perivascular lymphatic bed, but also the reverse transport of cholesterol from plaques to the liver, as well as the movement of cells containing cholesterol, oxLDL [518]. Thus, there is scientific interest in uncovering the significance of the adventitia zone in the regulation of cholesterol metabolism in the vessel wall.
Exogenous intervention using polysaccharide polymers in the process of atherogenic inflammation in the vessel wall and the formation of soft plaques can be represented as follows. It is known that the physical synthesis of chitosan and sulfated polysaccharides is accompanied by the self-assembly of polyelectrolyte complexes (PECs) and the formation of a nanoparticle core [544]. Nanoparticles can be formed by artificially grafting hydrophobic moieties into chitosan molecules or by mixing chitosan with polyanionic molecules [545]. Nanoformation of particles occurs by twisting of a linear polymer due to electrostatic interactions and hydrogen bonds [546]. The small size of chitosan is able to quickly penetrate transmurally into the intimate area and bind hydrophobic groups of free crystalline cholesterol from destroyed “foamy” cells, encapsulating it into a core of nanoparticles [547]. Such complexes become water-soluble and acquire the ability to be translated beyond the cells of the intimate zone of the vessel and its middle layer [546]. Along with the indicated mechanism of decholesterolization, there is an active release of residual cells from cholesterol with the formation of reverse transmembrane flows of cholesterol under the endothelial membrane as part of the core of the chitosan nanosystem [548].
Such active reverse transport of cholesterol from viable macrophages into the interstitial space and blood plasma is due to the safe and specific delivery into cells of a number of key micro-RNAs in the composition of polymer nanoparticles [549]. Translation of cholesterol in the intimate zone and the middle layer of the vascular wall can also occur with the flow of monocytes. Such flow beyond the outer shell of the vessel can be provided by an electrostatic gradient, which is created due to the polycationic or amphoteric properties of the polysaccharide polymer [547]. Based on these findings, the selected formulation (F2) was further coated with chitosan and showed a marked increase in vesicle size (298 +/- 3.56 nm), a positive zeta potential (+17 mV), superior encapsulation efficiency (88.1 +/- 1.48%). Thus, artificial minimally invasive dislocation of chitosan nanoparticles in the fascial sheath of great vessels may mean a local reverse of the cholesterol mass and is designed for a directed flow of lipid mass towards the adventitia [550,551]. The extraction of cholesterol from macrophages can be carried out into HDL particles for translation into the bloodstream, as well as independently by foam cells or through the mechanism of efferocytosis [552,553].
The removal of cholesterol from the vessel wall should change the nature of the interaction of molecular markers in the area of atherosclerotic vessel damage. The drainage effect of the biopolymer reduces the inflammatory response and explains the morphological preservation of tissue in the dislocation zone of the gel structure, despite the close proximity to the systemic destructive process [554]. This idea needs confirmation and detailed analysis of the dynamics of proatherogenic and antiatherogenic markers in experimental animal models.
The presence of stationary proteins in the ECM and their active interaction with growth factors during translation to the cell membrane has shaped research strategies for isolating GF s in order to protect their activity and more efficient presentation, significantly reducing the dose of angiogenic protein required for biological effects [555]. In research in It has been convincingly shown in vitro that in the case of adsorption of a non-linear, but a globular form of the ECM protein onto a polymer base, the attachment of growth factor (VEGF) does not occur, and, therefore, there is no active presentation of the complex to the cell membrane [556].
The use of three-dimensional media as a delivery system and presentation of growth factors to cells: ECM proteins, growth factors and a supporting structure in the form of a polymer is a promising direction. Growth factor-associated scaffolds generally induce more stable vasculature. High-quality studies have established that a multicomponent delivery system of angiogenic growth factor, for example, VEGF, consisting of fibronectin (FN) with adsorbed VEGF and polyethylene acrylate polymer (PEA), is capable of not only in vitro, but also in vivo create the phenomenon of cell stimulation at the highest level as a result of the presentation of growth factors in close proximity to integrin binding sites [556].
The low efficacy of angiogenesis stimulation endpoints in clinical settings may be due to the lack of a precise route of administration of a water-soluble neutral or weakly alkaline transport system [425]. Polymer matrices are proposed as a transport system in experimental models [557]. For example, when bFGF is included in a polymer delivery system (alginate capsules), the authors obtain positive angiogenic results [558,559]. Therefore, it is necessary to propose alternative spatiotemporal strategies for delivering growth factors to the ischemic zone.
Along with the very positive results of the analysis of the mechanisms of action and activation of angiogenesis in experimental animal models of ischemia, the insufficient effectiveness of the use of growth factors in patients in the clinic defines problems and sets the task of solving them. Priority solutions should include modification of the delivery system of regulatory proteins through the ECM for specific presentation to the cell surface. The most realistic solution may be the production and modification of natural biomaterials with high compatibility with ECM and GFs, which can be used both in combination with stationary ECM proteins and independently. Physical or chemical encapsulation of growth factors in a transport system should preserve the activity of proteins during natural degradation. Delivery of target molecules as part of a hydrogel matrix to the ischemic zone should quickly cover a large area of the arterial bed and come into contact with cells and affinity structures in the adventitial, middle and intimal zones of the vascular wall. For a detailed analysis of the mechanisms of action of implants of various origins, it is important to use animal models in which the process of ischemia develops gradually and over a long period of time with the necessary control series of experiments. The use of heparin or heparin-like structures in the delivery system is a good idea to solve the issues of creating an effective system for transporting growth factors and their sustainable release in the ischemic zone [4].
Problems of Angiogenesis Activation
Thus, the task of future research is to elucidate the mechanisms of impaired angiogenesis in the ischemic zone of the wall of the main vessel and the mechanisms of recovery using modern technologies for obtaining and using a delivery system for growth factors, reconstruction of the endothelial-mural apparatus of the intimal- subintimal zone of the artery affected by atherosclerosis.
Experimental and clinical work on therapeutic angiogenesis has used the strategy of endogenous modulation of both endoangiogenic growth factors (FGF-1,2 and VEGF165) and their genes (VEGF165, VEGF121, VEGF189 and FGF-5), as well as the administration of recombinant exogenous drugs. Their effect has been studied in patients with vascular ischemia of the lower extremities and in models of hindlimb ischemia in small and medium-sized rodents and models of acute and chronic myocardial ischemia in dogs and minipigs [560-564]. Scientific studies have sought to confirm or deny the mitogenic effect of VEGF on the vascular endothelium [565]. And, nevertheless, in almost all cases, the introduction of a growth factor or its gene stimulated the development of collaterals and new capillaries without their regression after the cessation of the introduction of growth factors.
When planning research projects aimed at obtaining and regulating angiogenesis in ischemic tissues, the authors aim to use artificial delivery systems capable of sequential and spatiotemporal presentation of GFs in the ECM. In this case, the efficiency of exogenous loading or endogenous opsonization of GF s is of paramount importance of their bioactivity and bioavailability. The physical form of the delivery system itself, in the case of large-area vascular reconstruction, should be a solution or liquid gel capable of easily filling the ECM area [566]. If the reconstruction of tissues included in a large area of the blood supply, where the diffusion limit exceeds 200 μm, is to be done, then the task of creating a functional network of blood vessels becomes necessary [567]. At the same time, researchers are focusing their attention on obtaining liquid intelligent hydrogels with the inclusion of growth factors with different functions, as well as filling a biodegradable structure with a pre-formed vascular network [17,568]. The development of such injectable vascularized hydrogels with improved adaptation to the affected area, reaching deep tissues with minimal invasiveness and acting as carriers for GFS and cells represents a significant step forward in the development of regenerative medicine [569]. This is especially important for large tissue structures where diffusion alone is not sufficient.
Creating combinations of angiogenic matrices will promote cell adhesion and increase the coordinated expression of growth factors. Such an algorithm will provoke the formation of endothelial networks and provide a multicomponent system of signals that implement vascularization [6,570-572]. Researchers can pre-incorporate the creation of these signaling pathways into the designs of smart hydrogels.
The main problem limiting the use of growth factors to activate angiogenesis is the lack of conditions for prolonged and stably active elution of protein molecules in the inflammatory zone. Proteolytic degradation of growth factors significantly reduces their half-life [573,574]. Rapid diffusion of water-soluble growth factor from microspheres or nanoparticles can be overcome by incorporating an intermediate transport system into three-dimensional liquid or semi-solid hydrogel matrices [575-578]. It has been established that the inclusion of growth factors in a polymer carrier improves the effect of proteins on cells due to the preservation of the protein structure from enzymatic hydrolysis [579]. However, problems in creating a system for transferring growth factors may also arise from blocking interaction of growth factors with each other, as well as from the interaction of the matrix itself with encapsulated growth factors.
As a protective delivery system for the active form of a growth factor, such as VEGF, researchers used a poly(lactic-glycolic acid) gel, which retained the factor's activity for a long time in a phosphate-saline solution upon its release [580]. At the same time, the simple technology of mixing the growth factor with the polymer before the gelation process is attractive. Despite the low stability and uncontrolled elution of growth factors in the delivery system after physical synthesis, careful selection of the polymer backbone for the delivery system should be considered [581-583]. Covalent immobilization of a growth factor upon contact with tissues will mean degradation of the complex at a rate corresponding to the rate of enzymatic or hydrolytic cleavage of the chemical bond [584]. In this case, growth factors may lose their biological activity due to steric hindrance in binding to GFR as a result of the closure of active centers during the immobilization reaction [585]. Thus, dosed and long-term delivery of growth factors to cells requires the inclusion of proteins in a reliable delivery system in the form of smart polymer matrices with a known degradation characteristic [557,579,586]. Moreover, the inclusion of matrices in reconstructed tissues means an addition to ECM. Each growth factor has a different degree of affinity for the natural or synthetic carrier, which creates a controlled release rate [587,588]. Under these experimental conditions, low doses of growth factor (20 μg) can be very effective. Solving this problem will be the main goal of obtaining high activity of local angiogenesis. This approach can also solve the problem of toxicity of growth factors or their inhibitors, provided that high doses of protein are used [589].
Effects of Endothelialization of Polysaccharide Matrices in Atherosclerosis. The Role of Sulfated Polysaccharide Derivatives
Upon contact with living tissues, microvessels can grow from the host vasculature and penetrate into artificial scaffolds through natural process of angiogenesis. However, such neovascularization is not sufficient to cover the entire area of the implanted structures; it occurs over a long period of time, over several days or weeks, which causes irreversible changes in the cells that have penetrated the matrix [590]. Therefore, translation of the microvascular network within engineered tissue constructs requires organized angiogenesis.
Research laboratories are searching for natural materials and their combinations, hybrid materials that can cause self-assembly of blood vessels. These building polymer materials themselves have angiogenic properties, since they contain specific ligands that implement three-dimensional conditions for intercellular communication, cell migration and various growth factors to demonstrate high recovery rates. The unique properties of natural polysaccharides include not only the ability to biodegrade, but also have high biocompatibility, the ability to block protein- polysaccharide structures on the microbial cell surface, and provide transmembrane transport.
The emergence of dynamic hydrogels, which can quickly change their physical characteristics in response to cellular contacts, is another promising direction for creating structures with similar properties of natural tissues [591]. Early vascular endothelialization around the implant is a primary goal and will mean oriented sprouting of precursors or specialized cells, as well as signaling molecules of intercellular communication. Modern developments of hydrogels as injectable scaffolds for growth factor delivery systems show good prospects for stimulating angiogenesis. Such matrices are less invasive and easily take on a configuration in the implantation zone [592]. It should be added that endothelial cells are capable of forming vascular networks, often without the prior addition of specific signals or growth factors, but only as a result of implantation of a free matrix. This spontaneous organization of endothelial cells leads to the maturation and stabilization of vascular structures and is determined by an increase in the number of vessels with a lumen [593]. Vascular networks form both in the ECM and in the body of the implanted matrix [594-596]. A large number of viable cells integrate into the scaffold, grow into avascular areas of the scaffold and vascularize them. Certain types of endothelial cells with high proliferation, present primarily in the adventitia (circulating endothelial progenitor cells (EPCs), endothelial colony-forming cells (ECFC), may play a crucial role in the revascularization of new territory [597].
The hydrogel mesh of the frame over the entire porous surface requires stable and prolonged filling with endothelial cell mass. And inducing angiogenesis not only on the outer surface of the implant, but also in the internal pore space is of key importance. Earlier studies showed that this problem can be solved by local conjugation and subsequent long-term delivery of angiogenic growth factors from the surface of the pores of the construct [321,598].
Effective minimally invasive delivery of angiogenic constructs to the affected area to create a hierarchical organization of the native vascular network is one of the main goals of vascular engineering [6]. Incorporation of angiogenic growth factors with endothelial cells in fluid matrices can induce sprouting or self-assembly of endothelium to create stable and perfused vascular networks with an open lumen [599-602]. In such transfers, the time sequence of introducing a cocktail of growth factors into the regeneration medium is very important. The simultaneous presence in the environment of inhibitory angiogenesis factors, such as platelet- derived growth factor B (PDGF) and angiopoietin 1 (Ang1), and activating pro-angiogenesis factors, such as endothelial growth factor (VEGF) and angiopoietin 2 (Ang2), leads to inhibition angiogenic reaction. Therefore, determining the levels of certain regulatory molecules in the wall of a large vessel will indicate the direction of action of the implant and, indirectly, the state of tissue perfusion. For example, low levels of PDGF and Ang1 in the environment mean that the inhibitory effect is abolished in the presence of high levels of VEGF and Ang2.
It is known that close contact with animal tissues of an initially cell-free polysaccharide polymer matrix stimulates the synthesis of angiogenesis products and triggers the process of endothelialization of the implant. Upon contact with the maternal vascular network (for example, after implantation of the construct into the perivascular zone), induced proliferation of the vascular endothelium occurs and its translation into the intimate zone, which culminates in intensive new formation of microvessels. Under favorable conditions, this process affects the artificial matrix zone. This translation is accompanied by active synergistic formation of molecules of the VEGF and FGF families, as well as their genes, by different types of cells [41,44]. Under conditions of inflammation and the presence of the effect of hypoxia, neoangiogenesis factors (VEGF, FGF, HGF, angiogenin, angiopoietins) and nonspecific vascular growth stimulators (IGF-1, TGF, TNF, nitric oxide, IL 8, MMPs) become triggers for endothelial cells that have receptors to growth factors [40,603]. Thus, the initial start of endothelialization is important from the standpoint of the growth of additional vessels in the implantation zone, changes in the activity of signaling molecules, leading to improved blood circulation in tissues. This direction of action is important in the development of chronic ischemia of the lower extremities. Understanding the interaction of signaling molecules during the formation of atherosclerotic plaques and in the presence of a polysaccharide construct is the goal of many studies.
However, technological approaches for creating a vascular network do not fully meet the objectives of the natural hierarchy of vascular structure. The artificial organization of the vascular wall currently does not correspond to the dimensions of the vascular tube: large vessel diameters corresponding to arteries or arterioles do not meet the necessary requirements for high hemodynamic loads during tissue perfusion. In large-diameter vessels, the presence of a single-layer endothelium and the absence of a middle layer at high pressure and increased blood perfusion will disrupt the architecture and function of the created network. The design model for now remains self-assembled vascular networks with a more accurate copy of the native structure.
Naturally derived polymers are known to have innate biocompatibility and exhibit proper cell adhesion and cell- dependent degradation, allowing hydrogel remodeling in a manner reminiscent of natural tissue ECM. The use of hyaluronic acid (HA) for this purpose is designed to accommodate the high susceptibility of HA to chemical modifications, allowing the development of a hydrogel that supports cell adhesiveness and presents angiogenic factors.
The inclusion of growth factors in artificial hydrogels significantly increases the functional integrity of proteins and maintains high angiogenic activity [579]. It is known that porous heparinized chitosan or collagen-chitosan matrices containing growth factors showed a high level of angiogenesis compared to control series of experimental animals. For example, the elimination of wound skin defects and the favorable development of the dermal fibroblast population in the wound occurred against the background of sustained delivery of recombinant human granulocyte-macrophage colony-stimulating factor (rhGM-CSF) and high expression of VEGF and TGF-beta1. These findings confirmed the formation of progressive angiogenesis [604,605]. Thus, the creation of a system consisting of natural scaffolds, various cell sources, growth factors, cytokines and mechanical stimuli is aimed at solving the problems of vascular tissue engineering.
The use of a collagen-chitosan nanofibrous scaffold creates favorable conditions for adhesion, translation and proliferation of endothelial cells of the main artery and preserves their vascular phenotype [606]. It has been established that heparinized linear polysaccharides with multiple side chains are able to covalently bind various ligands, including growth factors, cytokines, and protect protein molecules from proteolysis [607].
The migration of cells and their growth factors in the mural zone is ensured by a family of proteases and matrix metalloproteinases that cleave delivery systems and release deposited growth factors into the environment. In addition to donating active molecules along their path through the ECM, linear polymers can present growth factors to their receptors using the sulfhydryl group as a cofactor. There is an assumption that during the gradual degradation of, for example, sulfated chitosan implanted into the tissues of the vascular system, the matrix opsonized by growth factors will release soluble GFs into the medium, for example, FGF and VEGF isoforms, which can be released from the linear polysaccharide by enzymatic hydrolysis.
The role of incorporation of sulfated polysaccharide into a porous polyethylene glycol hydrogel was demonstrated in [608]. It has been shown that implantation of a chitosan-heparin-polyethylene glycol complex into tissue increases the efficiency of delivery of vector molecules into cells and significantly increases the level of angiogenic factors in this construct. The analysis shows good filling of the resulting transport system with factors such as sonic hedgehog (Shh) and vascular endothelial growth factor (VEGF). If the direct goal is to fill the hydrogel matrix with vascular endothelium, then researchers use a structure containing microchannels. For example, the formation of microchannels with an active surface based on highly deacetylated chitosan creates a bed for endothelialization of the framework. If such microchannels are loaded with angiogenic growth factor, then active adhesion of the vascular endothelium occurs. The high degree of deacetylation of the chitosan matrix plays an important role in the degree of endothelialization.
Direct contact with the wall of vessels of a polycationic or polyanionic hydrogel structure based on the sulfated form of chitosan, chitosan ascorbate, chitosan hydrochloride, sodium salt of alginic acid leads to proliferation of the vascular endothelium [609,610]. This endothelium surrounds the polymer matrix and stimulates lateral budding of the vessel wall. Such structures in fascial vascular sheaths create the effect of therapeutic angiogenesis. It was shown that endothelial cells reprogrammed from embryonic stem cells, encapsulated in chitosan hydrogel in combination with VEGF, when locally introduced into tissues of animals with a model of hind limb ischemia, led to neovascularization through the mechanisms of vasculogenesis and angiogenesis with effective restoration of blood flow in ischemic tissues [611].
In the polymer strategy for angiogenesis, researchers currently prefer to use complex hybrid hydrogels, consisting of several natural, synthetic or semi-synthetic polymers, several growth factors and cell types [612,613]. Since many models of critical limb ischemia (CLI) corresponded to acute circulatory disorders and were performed on small animals (mice and rats), the objectives of angiogenesis were, first of all, local delivery of the construct to the ischemic zone and prolonged elution of growth factor immobilized on the surface of the carrier, or from a deep layer of a polymer network [611,614]. Despite the active formation of the vascular network in the delivery zone of the hydrogel structure, the lesion area, as a rule, significantly exceeds the area of presence of the matrix with growth factors. The distance between individual vascular networks exceeds the possibility of their contact with each other [615]. The problem is the lack of technology for minimally invasive delivery of the construct to a large affected area, for example, with atherogenic inflammation. The hydrogel matrix, either as a standalone structure or containing stationary ECM proteins with growth factors, should be present over a large area of the affected tissue and have wide contact with growth factors, endothelial cells and structures of the mural zone.
Technologies for improving blood circulation in ischemic tissues are always accompanied by two final results: on the one hand, increased influx of cholesterol fractions into the vessel wall and, thus, the progression of the formation of lipid plaques, on the other hand, improved blood circulation in surrounding tissues. For example, a cytokine such as IL-20 and its receptors are expressed in human atherosclerotic plaques and in mice with apolipoprotein E knockout. This means the progression of atherosclerosis. However, in this case, active direct and indirect induction of angiogenesis occurs, which is regarded by researchers as an important proatherogenic mechanism of action of IL-20 [168,169]. Therefore, one should be aware that systemic stimulation of angiogenesis against the background of an atherosclerotic process can negatively affect the final results of therapy.
Therefore, one of the main challenges in this area is the development of improved methods for introducing, maintaining and measuring vascular perfusion after implantation of a delivery system of natural, synthetic or hybrid polymer constructs.
Artificial placement of a delivery system for cholesterol and LDL affinity ingredients in a wide area of atherosclerotic vascular lesions can affect not only the intimate and middle layers of surrounding vessels with the normalization of cellular and intercellular structure, but also cause active angiogenesis. For example, preliminary implantation of protonated gel forms of chitosan, as well as its sulfated form, in rats and rabbits on a cholesterol diet into the perivascular canal (case) of large vessels of the limbs leads, on the one hand, to a significant local extraction of total lipid fractions from the wall of the femoral artery and triglycerides over a large extent of the vessel, on the other hand, to the morphological reconstruction of the vascular wall, the formation of a large number of microvessels in the adventitia zone and an increase in arterial perfusion of tissues in the affected area [387]. The creation of electrostatic and concentration gradients in the wall of the main vessel using a polysaccharide polymer will lead to spatially oriented elongation and branching of endothelial cells [616,617]. In addition, the introduction of a hydrogel matrix into the fascial canal under light pressure creates local tension, which can form an angiogenic microenvironment in the tissue. It is known that a change in the compression modulus of the hydrogel (in this case, between the walls of the case and the vessel itself) leads to the effect of endothelial growth into the matrix [615].
In this case, the strategy of forming vascular structures by direct introduction of a hydrogel matrix of cells, growth factors, or a combination thereof, into hollow channels previously prepared using known technologies, cannot be ruled out. The researchers expect that the channels will be perfused with tissue media and can be successfully seeded with endothelial cells. Cells can migrate from the channels into the matrix walls [618,619].
The high effect of new formation of the microvascular bed at the site of implantation of chitosan constructs is also confirmed in an intact animal, amounting to an increase in the number of microvessels after 30 days of the post-implantation period of 85- 96% [620]. The proposed concept of morphological restructuring of the qualitative and quantitative characteristics of vascular walls under the influence of natural polysaccharides requires an analysis of the molecular mechanisms of angiogenesis when attempting to intervene in the complex cascade of formation of a soft atherosclerotic plaque [516,609,620,621]. The administration of sulfated chitosan as a polyanionic polymer can provide a gradient of movement of growth factors and cofactors in the ECM to activate their receptors on neighboring cells. Binding of GF to a specific receptor initiates a signal to a transduction cascade with phosphorylation of intracellular domains, activates kinases, and subsequently transmits signals to generate a cellular response.
It is important to include in this analysis events in the adventitial and paraadventitial zones, which are interested in direct contacts with the delivery system of lipid affinity ingredients [496].
It was assumed that implantation of a polysaccharide hydrogel along the fascial canals along the arterial trunks will lead to widespread contact of the matrix with the elements of the created second additional ECM, located in the adventitia and beyond, in other words, ECM-2. Active affinity opsonization of the hydrogel by growth factors in the adventitial ECM -2 and simultaneous degradation of the water-soluble and hydrophilic matrix with delayed elution of GFs into the environment will lead to the formation of adventitial vasculature [622,623]. In this case, the advenitial vascular network, formed on the basis of polymers of natural origin, will have innate biocompatibility, exhibit the necessary cell adhesion and cell-dependent degradation. In this regard, based on the used exogenous hydrogel, one can expect its remodeling, which in composition imitates living ECM. This idea is reminiscent of the design of decellularized extracellular matrix (dECM) obtained from organs and tissues, which has the characteristics of an ideal tissue scaffold with a complex composition, vasculature and a unique tissue-specific architecture. It is assumed that biologically active molecules, such as growth factors and cytokines, can be stored in the decellularized matrix. However, there are doubts about maintaining the activity of growth factors, since the decellularization process takes a long time using toxic compounds, and the period of activity of growth factors is short. In addition, when using exogenous dECM, it is necessary to perform a number of manipulations and control the effectiveness of ridding the ECM of antigenic material; the decellularization process leads to the loss of the endothelial cell layer by the scaffold, which is critical for tissue regeneration. Such difficulties can be eliminated by the formation of endogenous ECM based on natural polysaccharides and a three-component cellular system from the adventitia. Calculations are made for the active flow of hydrogel around vessels of various calibers when the structure is implanted into the fascial sheaths of vessels and, thus, modeling the vascular tree according to a natural pattern. This approach is designed for complete endothelialization of newly formed blood vessels growing from the adventitial layer (Figure-scheme 8, Figure-photo 9I,10J,11KL).
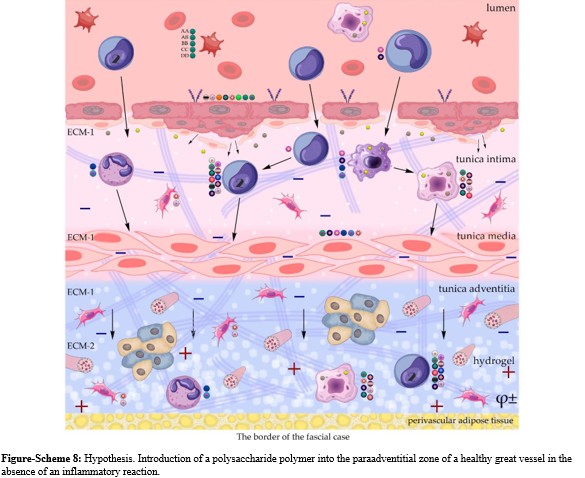
Activation of the endothelium with the expression of VEGF, FGF, active proliferation of endothelial cells, their migration to the subendothelial zone with simultaneous detachment of pericytes and activation of PDGF, recruitment of pericytes, their creep onto the new single-layer endothelium with sealing of their contacts, formation of the lumen of microcapillaries (ANG-1). Creation of permanent drainage of the subendothelial and middle zone of the main vessel due to the presence of concentration (C %) and electrostatic (ÃÃÂ?ÂÂ? ±) gradients in ECM-1 and ECM-2 using an amphoteric biopolymer. Such gradients attract monocytes from the bloodstream, form a pool of M2 type macrophages in all zones of the vascular wall, and ensure directed migration into the adventitia and beyond. As a result of productive inflammation in the paraadventitial zone under the influence of the polysaccharide polymer and expansion of the adventitia zone due to degradation and reorganization of the inflammatory reaction in the internal layers of the main vessel, there is no inflammatory reaction. Active angiogenesis and the formation of vasa - vasorum occurs in the adventitia and ECM-2.
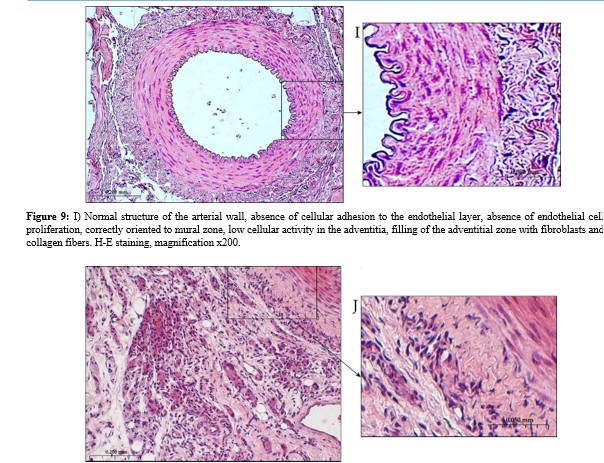
Figure 10: J) Cross section of the artery wall of a healthy rabbit. Presence of antiatherogenic productive inflammation in the paraadventitial zone of the artery of a healthy rabbit 1 month after implantation (1% gel of (ON-(2- sulfoethyl)chitosan, 60% SD (sulfation degree), 257 kDa of structural unit, 92-95% DD (deacetylation degree). Massive formation of new capillaries in the paraadventitial zone. Staining G-E. The thin histological sections (3–4 μ m) using a Leica automated system (Germany). Microphotographs were obtained using an Axio Imager A1 microscope with an AxioCam MRc5 photography system and an Axio Vision software (Carl Zeiss, Germany). H-E staining, magnification x200.
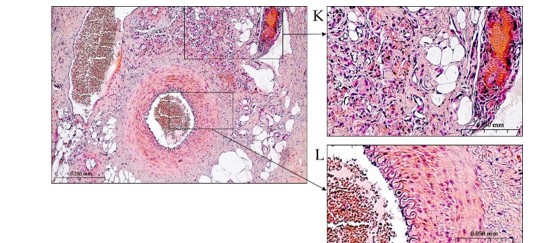
Figure 11: K) Formation of a productive inflammatory reaction outside the adventitia, L) Formation of a productive inflammatory reaction outside the adventitia. H-E staining, magnification x100
In addition, gel constructs implanted into the fascial bed are in close contact with adipose tissue, which can determine the differentiation of hematopoiesis and stimulate the regeneration of blood vessels from the adventitia layer. This idea can be based on the results of studies devoted to the co-cultivation of the main cellular elements of the vascular wall: vascular endothelium, VSMC and macrophage mass in vitro under the condition of sequential layering of some cells on others in the presence of a chemotactic agent [624]. It is important that all three cellular components are present in the adventitia in contact with the hydrogel and create a natural homeostasis that has the ability to perform organ-specific functions [625]. Moreover, it is known that the ideal cell types for hydrogel cellularization are endothelial cells [626].
It is assumed that such a hydrogel system will be additionally activated by the influx of LDL due to the transmigration of macrophages, as well as independently as a result of the functioning of the electrostatic gradient of the hydrogel matrix, which also has a high affinity for LDL cholesterol. From the perspective of the idea of reversing the atherogenic process, the attraction of macrophages loaded with cholesterol to the advenitial layer as a possible therapeutic target for reducing atherosclerotic inflammation is of great interest [627].
Despite the independent preparation of dECM with subsequent filling with cells, there is a noticeable active creation of hybrid hydrogels, which include additional materials of a polysaccharide nature that improve the entire structure during implantation (polycaprolactones, chitosan, alginate).
As mentioned above, when using copolymers obtained as a result of physical or chemical synthesis to activate angiogenesis, researchers use sulfated constructs that have a high capacity for growth factors. For example, the addition of heparin sulfate to chitosan leads to an angiogenic effect in tissues due to the immobilization of proangiogenic growth factors on the copolymer [628-631]. This effect is observed primarily due to the immobilization of proangiogenic growth factors on heparin. This immobilization is temporary and has a delayed release into the ECM [11,632-634]. As a result of long-term dislocation of heparinized polysaccharide in the tissue, a large number of microvessels are fixed in the ECM in place of the dissociated polymer.
Heparin has a high specific affinity for the FGF family, which has a heparin binding site in the form of numerous positively charged amino acids [635]. The higher the degree of binding of heparin on chitosan, the higher the capacity of the immobilized protein. However, it turns out that this effect occurs every time if chitosan is present in the design [636,637]. It was found that the inclusion of FGF in a chitosan-based hydrogel in the absence of heparin also revealed a stable release of growth factor in the area of chronic inflammation and stimulated the activation of endothelial cells (CD 34+) and fibroblasts in the walls of capillaries [638,639].
If a polylactone type construct is used, then the inclusion of heparin in it will also lead to an increase in the sorption of angiogenic growth factor [640]. Electrostatic interactions between positively charged amino acid residues of lysine and arginine, such as fibroblast growth factor (bFGF), vascular endothelial growth factor (VEGF), transforming growth factor-β (TGF-β), platelet- derived growth factor (PDGF), and negatively charged with 2-O -sulfate and N -sulfate groups of heparin not only increase the binding affinity, but also serve as a stable delivery system to the inflammatory zone [641-644]. In this case, the low stability of physical adsorption of heparin on a biodegradable matrix can be overcome by chemical modification, using, for example, copolymerization of gel matrices with different molecular weights. In this particular case, the initial incorporation of heparin into the low molecular weight poly(lactide-co- glycolide) copolymer (H-PLGA) and subsequent mixing with high molecular weight PLGA completes the attachment of the high molecular weight substrate and thus prolongs the elution effect, and hence the release of bound bFGF. Covalent conjugation between heparin carboxyl groups and surface exposed amino groups on PLGA scaffolds and subsequent physical attachment to the resulting bFGF complex led to prolonged desorption of growth factor into a medium with high biological activity against human umbilical vein endothelial cells (HUVEC) and the formation of blood vessels in the subcutaneous pouches of experimental mice [640]. Slow and controlled release of growth factors from scaffolds, such as platelet-derived growth factor BB (PDGF-BB), has been found to be important for high angiogenic outcome. In this case, an equally important result is the long-term preservation of the biological activity of the growth factor [645].
In earlier studies, the authors immobilized angiogenic factors onto an anionic polysaccharide such as sodium alginate. Rapid gelation, solubility in aqueous media, very high biocompatibility, and the resulting negative charge of the polymer molecule have enabled the preparation of hydrogels for the transport of biologically active signals or cells in animal models of CLI. For example, adding immune T-cell culture medium factors to the system and local implantation of the system into the ischemic zone significantly increased angiogenesis in the surrounding tissue [646].
It is known that alginate is used as a carrier of proteins to protect them from degradation, including for the delivery of growth factors through the ECM compartment [647,648]. To enhance angiogenesis and endothelial differentiation, lginate is widely used after combination with factors such as VEGF and bFGF. If you include VEGF in a sodium alginate molecule, you can obtain dosed elution of the growth factor and the formation of new capillaries [636]. It was established experimentally that rapid local sequential delivery of VEGF in high and low doses to ischemic tissues had the best therapeutic effect in the CLI model. Moreover, multiple local administration of VEGF created a better therapeutic effect [649]. The use of a combination of VEGF-loaded microspheres and hydrogel confirms the prolonged release of growth factor from the system into ischemic tissues [650]. If microspheres are included in the alginate gel poly(lactic-glycolic acid), then the property appears to control the release of angiogenic factors, for example, bFGF [598].
Considering that pure alginate initially has a low ability for cell adhesion due to the resulting negative charge, to increase the adhesion force, researchers create interpenetrating hydrogel networks by introducing polycationic polymers [651,652]. Mixing oppositely charged polyelectrolytes leads to the formation of complex coacervates-spherical droplets that are capable of non-covalently incorporating proteins and transporting them into the ECM [653-659]. In this case, the physical synthesis of the copolymer can provide a natural rate of degradation of this transport system, which can be controlled. The rate of ion outflow into the environment creates slow degradation of the matrix, which is very important for creating conditions for active angiogenesis in the ischemic zone. It has been established that the use of polysaccharide polymer matrices of cation-anion properties enhances the efficiency of delivery of vector molecules into cells and leads to overexpression of angiogenesis molecules, enhances endothelial recruitment and new formation of blood microvessels [608].
The inclusion of a heparin group in alginate significantly slows down the rate of degradation of the new construct, and at the same time, ensures a prolonged release of growth factors from cells encapsulated in the gel due to improved mechanical properties of the matrix [660,661]. The same effect can be created by mixing other sulfated molecules with alginate, such as cellulose sulfate or dextran sulfate [662,663]. Liquid hydrogels show promise in the form of simple injections to transport growth factors or ECM proteins to target cells to induce activation signaling pathways [664].
It is important to note that the aggregation state of the alginate construct can be changed by increasing or decreasing the pH of the gel. For active transfer of growth factors through the ECM in a liquid gel, it is enough to obtain a solution with a pH of 7.0- 7.4. The inclusion of cationic polymers in such a gel also acquires antibacterial properties, which is very important when introducing scaffolds for the purpose of tissue engineering [665].
If an acidic mucopolysaccharide, such as a hyaluronic acid salt, is included in the growth factor or cell transport system, a similar angiogenic effect can be obtained in the CLI model. Endothelial cells included in this hydrogel can be used to improve the condition of the ischemic zone by stimulating angiogenesis and arteriogenesis [666]. The addition of hyaluronic acid gel to the collagen matrix significantly affects the migration and differentiation of endothelial cells [667].
The natural polymer dextran can also be modified with various functional groups and cross-linked to form a biocompatible hydrogel scaffold. This nontoxic polysaccharide, consisting of linear α-1,6-glycosidic D-glucose linkages, is capable of stimulating angiogenesis [668]. The ingrowth of tissues into the matrix depends on the cross-link density and the degree of polymer swelling [306].
Elements of extracellular matrix (ECM) introduced into the hydrogel are able to recruit more endothelial cells, improve the local condition of the muscles of the ischemic limb and increase tissue perfusion for a sufficiently long time [669]. The positive effect of ECM cells indicates the importance of each element in the angiogenesis system in the vascular wall zone [670]. The same pattern was confirmed by an increase in fibroblast growth factor (FGF) capacity in alginate or collagen constructs when heparin was included [598,671,672]. The longer the elution from the growth factor delivery system takes place, the higher the density of the newly formed capillaries. In this case, the matrix can have either a solid or hydrogel consistency [673].
Among the polysaccharide polymers that can interfere with the cascade of molecular disorders during tissue ischemia, poly(lactic acid-co-glycolic acid) (PLGA), a copolymer of glycolic and lactic acids, has attracted scientific interest. The polymer is a biodegradable material with excellent biocompatibility that enables the technical fabrication of micro/nanoparticles or 3D scaffolds for biomedical applications. The use of this polymer as a loaded hydrogel in a mouse model of critical lower limb ischemia showed a high level of growth of proangiogenic factors and a decrease in the degree of tissue damage [674].
Thus, despite advances in the field of tissue engineering, numerous studies in in vitro vitro usually do not achieve adequate translational efficiency of the transfer system. Nevertheless, scientific interest in technological developments with detailed analysis of the mechanisms of angiogenesis, vasculogenesis and arteriogenesis will grow. Efforts to create functional prevascularized scaffolds with complex structures and material compatibility will lead to the creation of vascularized constructs suitable for clinical use. At the same time, the idea of forming such endogenous structures using regenerative capabilities in vivo remains very promising.
Induction of angiogenesis in ischemic tissues will be the most promising task in the treatment of early and late vascular patency disorders. Local reconstruction should take a central place in vascular engineering. Modeling a physiologically dynamic environment will play an increasingly important role in promoting natural maturation of the vascular system. The creation and use of hydrogels with different active molecular loads and cell mass is a very promising direction. The focus of scientific work should be related to the production and testing of hybrid polysaccharide polymers, as the safest and most responsive to the physiological needs of damaged tissues.
The development and production of new systems for delivering active molecules to the vascular lesion area requires, using an effective delivery route, to determine the comparative therapeutic effectiveness in the CLI model. On the same living object on both hind limbs at the same time, it is desirable to conduct a parallel analysis of the angiogenic effect of the hydrogel with determination of the levels of a wide cocktail of molecular markers. It should be expected that setting the goal of obtaining a picture of angiogenesis over time will significantly complicate the task. Apparently, the use of polymers of a polysaccharide nature in research, despite a number of their removable disadvantages, will receive priority, since they largely correspond to the structure and reaction of normal tissue under the action of internal and external stimuli, as well as the fact that a number of polysaccharides, especially with sulfated domains, has its own unique ability to independently regulate angiogenesis without additional load. The ability of sulfated hydrogels to self-opsonize growth factors when implanted into the ECM could likely be exploited in vivo without their preliminary encapsulation under in conditions in vitro, where preparing modified versions of growth factors for opsonization in a hydrogel can pose technical difficulties given the short half-lives of regulatory proteins [255]. In order to increase the efficiency of delivery of biomaterial to the zone of chronic inflammation, it will probably be necessary to reconsider the routes of introduction of hydrogels and, accordingly, their composition of growth factors. Intramuscular multipoint injection of a solution or hydrogel, intravenous or intraarterial administration of water-soluble growth factors do not always correspond to the calculated characteristics and are highly invasive manipulations. Attempts to introduce hydrogel and angiogenic products into suspected ischemic areas in chronic inflammation of the great arteries are not accurate and do not cover the vessels over a large extent. Recently published works devoted to the reconstruction of large arteries of the lower extremities in experimental atherosclerosis offer a minimally invasive technology for local delivery of hydrogel throughout the affected vessel of the hind limb of an animal (rat, rabbit) [387,516,609,620,621]. A number of studies have convincingly shown that early initial forms of ischemic damage to the vessels of the extremities and the formation of soft plaques in an animal model can be completely eliminated and the mechanisms can be started innate potential for revascularization and self-healing [539- 541]. The effect of decholesterolization of the wall of the great vessel and degradation of soft plaques over a long period after intrafascial implantation of sulfated chitosan hydrogel, as well as cationic polysaccharides, allows planning research to decipher the mechanisms of reverse development of local atherogenic lesions [387]. An optimized microenvironment of angiogenic ECM signals for the physiological presentation of morphogens can be achieved by introducing a sulfated unloaded matrix into the paraadventitial zone of the vessel, taking into account the active opsonization of the matrix by growth factors in vivo, which provides ideal conditions for cell migration, vascular invasion and progenitor differentiation. The combination in the adventitial layer, as well as in the hydrogel matrix, of endothelial cell precursors transmitted by the bloodstream and a mass of fibroblasts becomes very important for vascular tissue engineering.
The authors propose a hypothetical scheme for the regulation of angiogenic and proatherogenic growth factors in the wall of an arterial vessel during the correction of atherogenic inflammation using subfascial implantation of an amphoteric polysaccharide using the example of sulfated chitosan (Figure-Scheme 12, Figure- Photo 13MNO,14P,15Q,16S,17TU).
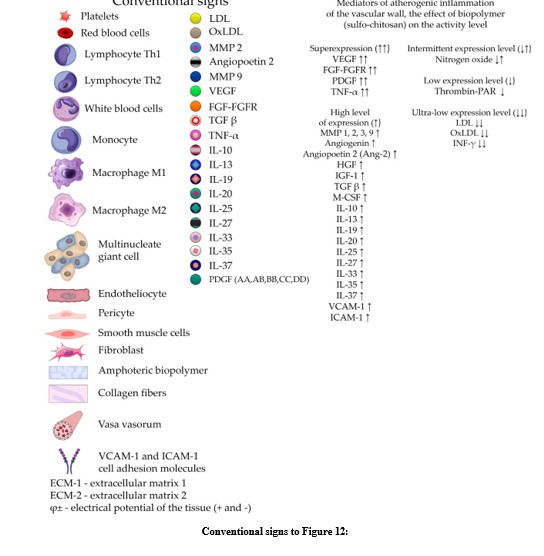
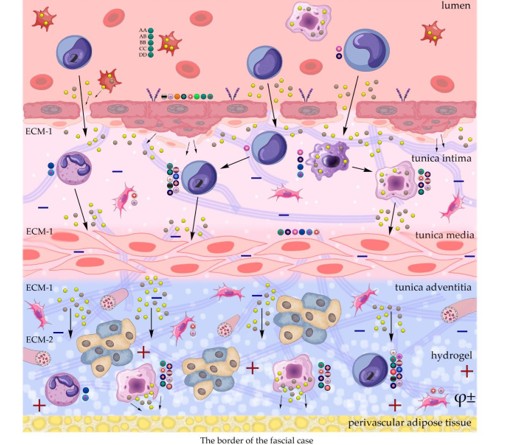
Figure – Scheme 12: Hypothesis. Significant expansion of the peri-adventitial zone, filling it with cell mass in close contact with the biopolymer. Streamlining the orientation of the layer of smooth muscle cells, a significant decrease in the number of “foam” cells, modification of macrophages into type M2, Th1-cells into type Th2, active reverse of cholesterol from macrophages and smooth muscle cells due to the high affinity of the biopolymer for cholesterol and LDL, suppression of apoptosis macrophages. The combination of active angiogenesis in ECM-1 in the intimate zone with the translation of leukocyte and macrophage mass into the area of adventitia and hydrogel matrix (ECM-2), the formation of a microvascular bed based on vasa - vasorum in the polymer biodegradation zone. The presence of proliferating macrophages in the giant multinucleated cells form that resorb the polysaccharide polymer. This process involves a wide basin of the main vessel due to the active flow of hydrogel around vascular branches of various calibers along their fascial sheaths. Thus, the hypothetical scheme is designed for widespread endothelialization of new blood vessels growing from the adventitial layer, active outflow of atherogenic inflammatory infiltrate from the intimate zone into the adventitia, translation of the vascular endothelium beyond the vascular wall into the tissue compartment, including into the degradable hydrogel layer.
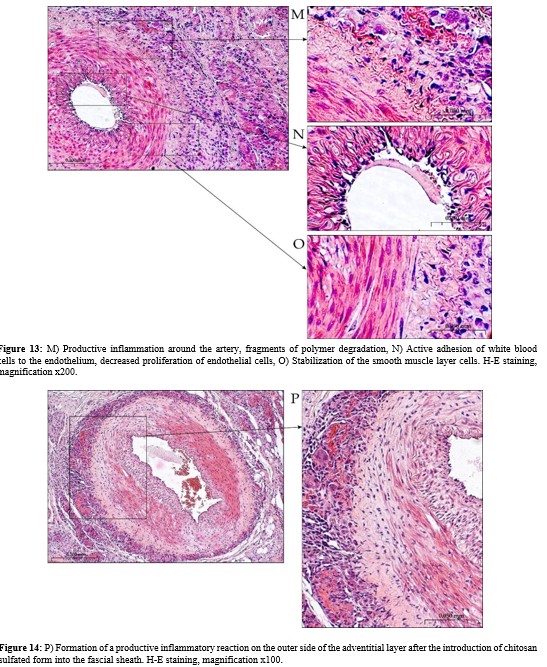
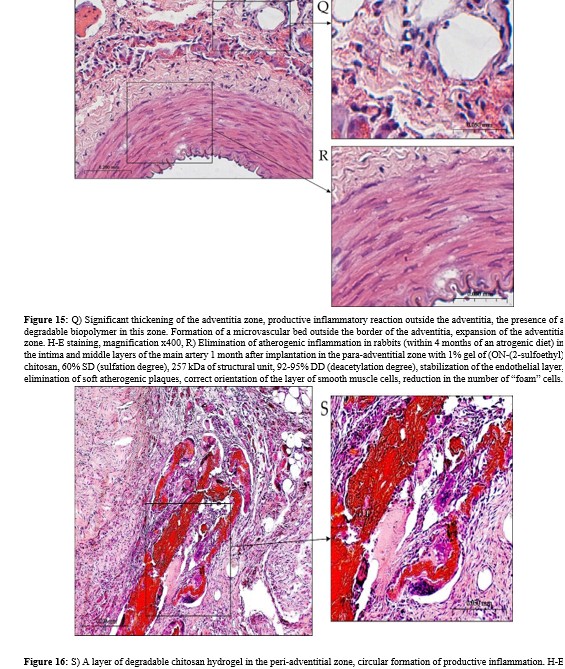
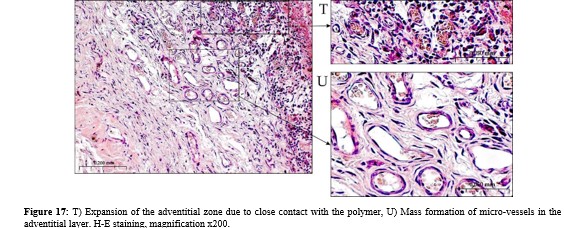
If fibrin gel loaded with this cellular system is preliminarily used, the resulting microcapillary network anastomoses with the recipient’s vascular network in a short time [675]. In addition, the introduction of the matrix itself occurs on a partially prepared platform, since the ischemic tissue is already partially vascularized, and implantation of the hydrogel matrix will be aimed at expanding the microvascular networks to promote remodeling of collateral arteries and restoration of physiological blood flow [676]. When endothelial cells are located in preformed microchannels, it has been confirmed that they are able to recruit into adjacent microchannels and establish cell-cell contacts in the implanted hydrogel [677-679]. If endothelial cells are able to travel short distances over a short period of time, then the creation of a vascular network in the area of a large vascular basin requires long-term dislocation of the implant with the presence of viable cells [680]. The end result of this outgrowth is the formation of a perfused vascular network.
Vascular diseases of any location always involve a long process. Acute circulatory disorders often do not require angiogenesis stimulation strategies. For this purpose, effective surgical correction methods are used. Ischemia of the tissue, in which the entire cascade of molecular disorders develops, corresponds to the chronic course of the disease. In this regard, multiple published models of acute tissue ischemia of the hind limbs in small laboratory animals do not fully meet the goals of stimulating angiogenesis and understanding the mechanisms of ischemia development. The most adequate animal models should be those in which the lumen of blood vessels gradually closes with the formation of an inflammation process. One of the most striking models of chronic inflammation is experimental atherosclerosis, which is successfully implemented in laboratory rats and, especially, rabbits. The model of cholesterolosis by Anichkov and Khalatov, developed on rabbits in 1913, is a striking example of obtaining a specific chronic inflammatory process that occurs over several months thanks to a special cholesterol diet [681]. Despite the presence in laboratories of strains of mice and rabbits with hereditary hypercholesterolemia, this model is in demand and is widespread.
Thus, modified lipoproteins and cholesterol crystals accumulate in the arterial intima and stimulate the formation of proatherogenic macrophages, foam cells and inflammation. Defective efferocytosis of apoptotic foam cells leads to the formation of a necrotic core, a sign of failure to resolve inflammation. Resolution of inflammation is mediated by specialized pro-resorption lipid mediators, proteins and signaling gases. Improving the balance between pro-inflammatory and anti-inflammatory factors contributes to the reverse development of local atherogenesis. Active direct intervention in the reconstruction of the adventitial layer of the arterial wall using cholesterol-affinity polysaccharide hydrogels containing a cocktail of growth factors creates conditions for the formation of additional extracellular matrix and reversal of cholesterol mass from the intimate zone. Using a hydrogel, creating concentration and electrostatic gradients in a large area of adventitia over a long artery can be one of the effective ways to degrade early soft atherogenic plaques and local restore the damaged structure of the vessel wall. Growing scientific interest in the previously insufficiently studied adventitia indicates its important role in angionesis and atherogenesis.
Conclusion
The Concept of The Mechanisms of Action of Sulfated Chitosan During Its Paraadventitial Implantation in A Model of Experimental Atherosclerosis
Various methods have been developed to couple growth factors with natural or synthetic biomaterials and chemicals. These immobilized factors will be available to cells that come into contact with the matrix, providing a highly localized signal to control cell fate. In the case of active angiogenesis, growth factors can bind to the surfaces of the scaffolds, providing proangiogenic signals to the surrounding tissue. [682]. Injectable scaffolds are a promising approach to promote angiogenesis as they are less invasive than implantation of solid scaffolds and can contour to fill cavities [683,684]. Growth factors can be active in a bound state or activated as a result of detachment from the matrix. Site- specific binding of growth factors and other biological molecules allows control of multiple growth factor functions and their delivery. There are two main strategies for direct presentation of growth factors on external matrices: a) physical adsorption due to hydrogen bonding of proteins; b) covalent immobilization of growth factor or growth factor mimic molecules into a matrix (covalent approach). Regardless of which technique is used to immobilize growth factors, scaffolding also imparts to cells the ability to recruit functional proteins into intimate cell-cell contact.
Increased activation of growth factor receptors, such as VEGF-A, BDGF or FGF-2, in the extracellular matrix is associated with the inclusion of integrin in the interaction chain. Chemical or physical modification of the surface of extracellular matrix substrates with heparin is used to bind growth factors through their affinity for grafted heparin. Heparin-based growth factor delivery systems have demonstrated the ability to provide sustained release of growth factors [685].
Cell migration, determined through the integrin-growth factor receptor system signaling, can be activated by an electrostatic gradient in the presence of sulfated chitosan.
High affinity adsorption of growth factors in the ECM to the implanted sulfated form of chitosan ensures the connection of growth factors with the ECM, and, therefore, stimulates unhindered diffusion to the surface of endothelial cells and cells of the mural zone. Provided that a growth factor adsorbed on the carrier, for example, VEGF, will be actively presented to endothelial cells at a low level of the Notch signal, the process of regional angiogenesis is expected to be high.
When cells come into contact with the implanted chitosan polymer, physically immobilized and carrier-bound growth factors will be available to the cells, likely providing a highly localized signal to trigger cell function. In this case, the role of intermediate structures that perform the functions of opsonization of growth factors on the surface of the transfer system can be played by both sulfhydryl groups of the polymer itself and oligopeptides, fibronectin, collagen, elastin or the glycosaminoglycan family. Such a complex multicomponent growth factor delivery system can significantly increase the survival of endothelial cells and restore local blood perfusion in ischemic tissue [686].
It is assumed that the polyanionic soluble form of sulfated chitosan, when implanted into the paravasal fascial sheath of the main vessel, will be non-toxic for growth factors adsorbed in the ECM. An important condition is the preservation of protein activity in the delivery system. The chitosan platform with a pH around 7 or slightly alkaline can modify the structure of the transport system, immobilize growth factors, increase the degradation time and release of growth factors into the ECM compared to many cationic polymers.
It is possible that the introduction of a chitosan hydrogel with sulfhydryl groups into the paravasal space leads to the formation of affinity complexes with cholesterol as stable and monodisperse gelation platforms that include growth factors [687].
The rate of degradation of the transport system probably depends on the concentration of the polysaccharide polymer [688]. It is assumed that changing the concentration of polysaccharides in tissues (implantation of sulfated chitosan into the paravasal zone of the main vessel) changes the dynamics of the release of growth factors from vascular endothelial cells, smooth muscle cells, other ECM cells, the regenerative profile of lymphocytes, and macrophages. In addition, implantation of a certain volume of polymer into the main vessel casing creates mechanical stress on the tissue. This exogenous factor stimulates the production or release of growth factors in the ECM and cell-cell contacts with activation through GFs receptors and integrin signaling pathways. The cell response manifests itself in the form of differentiation, proliferation and migration. The high affinity of polysaccharide polymers for cholesterol and LDL, active vascularization of ECM-2 provoke a gradient of cholesterol translation towards the hydrogel “shirt”, draining cells and lipids from the subintimal zone of ECM -1 to the ECM-2 zone. In addition, short chains of hydrolysis products of the polysaccharide implant fill not only ECM-2, but also diffuse back into ECM-1, binding to the cell surface of polymorphonuclear leukocytes and macrophages. It is assumed that such a supply of cholesterol from the subintimal zone can be combined not only with the translation of “foam” cells, but also of extracellular oxLDL , carried out of the membrane by the foam cells themselves using the ABCA1 and ABCG1 transporters. Cholesterol efflux plays an important role in antiatherogenesis, and manipulation of this process may provide a new therapeutic approach to great vessel pathology [689].
There is an assumption that activation of efferocytosis with the help of polysaccharide matrices, which reduces the mass of apoptotic cells, can prevent secondary necrosis and inflammation, reduce the growth of necrotic nuclei [690]. Resorption and active drainage into position of a polymer having an electrostatic charge are aimed at restoring the integrity and function of tissues during inflammation. Switching the function of pro-inflammatory macrophages to an anti-inflammatory pool of cells using specific lipids opens up the prospect of controlling the atherogenic process, especially at an early stage of development [381]. Obtaining a mass of non-inflammatory macrophages in the wall of an arterial vessel is a very interesting project [691]. This process ends with local decholesterolization and restructuring of the mural zone of the vascular wall. Such processes in the wall of the main vessel change its morphology and function, which should be reflected in improved perfusion characteristics. Bringing a gel matrix to the zone of ischemia and dislocation of atherogenic plaques as an independent structure for contact with stationary ECM proteins, endogenous endothelial cells and cells of the mural zone to activate angiogenesis is considered an effective treatment regimen. This is due to the fact that more than 90% of the exogenous cell mass is eliminated within 24 hours after transplantation and after 4 weeks it is only 1% [692]. In connection with this postulate, it is necessary to pay special attention to the mechanisms of the antiatherogenic action of the adventitia of large arterial vessels.
Developing mechanisms to improve the stability of encapsulated proteins and their release over time will require the efforts of researchers working in several disciplines, including chemical synthesis, experimental tissue engineering, pathology modeling, molecular biology and medicine.
References
- . Sing, P., Rudan, D., Zhu, Y., Fowkes, F. J., Rahimi, K., Fowkes, F. G. R., & Rudan, I. (2019). Global, regional, and national prevalence and risk factors for peripheral artery disease in 2015: an updated systematic review and analysis. The Lancet Global Health, 7(8), e1020-e1030.
2. Steffen, M. W., Undavalli, C., Asi, N., Wang, Z., Elamin, M. B., Conte, M. S., & Murad, M. H. (2015). The natural history of untreated severe or critical limb ischemia. Journal of vascular surgery, 62(6), 1642-1651.
3. Criqui, M. H., & Aboyans, V. (2015). Epidemiology of peripheral artery disease. Circulation research, 116(9), 1509- 1526.
4. Han, J., Luo, L., Marcelina, O., Kasim, V., & Wu, S. (2022). Therapeutic angiogenesis-based strategy for peripheral artery disease. Theranostics, 12(11), 5015.
5. Norgren, L., Hiatt, W. R., Dormandy, J. A., Nehler, M. R., Harris, K. A., Fowkes, F. G. R., & Tasc II Working Group. (2007). Inter-society consensus for the management of peripheral arterial disease (TASC II). Journal of vascular surgery, 45(1), S5-S67.
6. O’Connor, C., Brady, E., Zheng, Y., Moore, E., & Stevens, K. R. (2022). Engineering the multiscale complexity of vascular networks. Nature Reviews Materials, 7(9), 702-716.
7. Khademhosseini, A., & Langer, R. (2016). A decade of progress in tissue engineering. Nature protocols, 11(10), 1775-1781.
8. Riley, C. M., Fuegy, P. W., Firpo, M. A., Shu, X. Z., Prestwich, G. D., & Peattie, R. A. (2006). Stimulation of in vivo angiogenesis using dual growth factor-loaded crosslinked glycosaminoglycan hydrogels. Biomaterials, 27(35), 5935- 5943.
9. Chiu, L. L., & Radisic, M. (2010). Scaffolds with covalently immobilized VEGF and Angiopoietin-1 for vascularization of engineered tissues. Biomaterials, 31(2), 226-241.
10. Layman, H., Li, X., Nagar, E., Vial, X., Pham, S. M., & Andreopoulos, F. M. (2012). Enhanced angiogenic efficacy through controlled and sustained delivery of FGF-2 and G-CSF from fibrin hydrogels containing ionic-albumin microspheres. Journal of Biomaterials Science, Polymer Edition, 23(1-4), 185-206.
11. Roberts, J. J., Farrugia, B. L., Green, R. A., Rnjak-Kovacina, J., & Martens, P. J. (2016). In situ formation of poly (vinyl alcohol)–heparin hydrogels for mild encapsulation and prolonged release of basic fibroblast growth factor and vascular endothelial growth factor. Journal of Tissue Engineering, 7, 2041731416677132.
12. Zieris, A., Chwalek, K., Prokoph, S., Levental, K. R., Welzel,P. B., Freudenberg, U., & Werner, C. (2011). Dual independent delivery of pro-angiogenic growth factors from starPEG- heparin hydrogels. Journal of Controlled Release, 156(1), 28-36.
13. Yancopoulos, G. D., Davis, S., Gale, N. W., Rudge, J. S., Wiegand, S. J., & Holash, J. (2000). Vascular-specific growth factors and blood vessel formation. nature, 407(6801), 242- 248.
14. Li, B., & Xiu, R. (2013). Angiogenesis: From molecular mechanisms to translational implications. Clinical hemorheology and microcirculation, 54(4), 345-355.
15. Jansen, P. L., Rosch, R., Jansen, M., Binnebösel, M., Junge, K., Alfonso-Jaume, A., ... & Mertens, P. R. (2007). Regulation of MMP-2 gene transcription in dermal wounds. Journal of investigative dermatology, 127(7), 1762-1767.
16. Pober, J. S., & Sessa, W. C. (2007). Evolving functions of endothelial cells in inflammation. Nature Reviews Immunology, 7(10), 803-815.
17. Brudno, Y., Ennett-Shepard, A. B., Chen, R. R., Aizenberg, M., & Mooney, D. J. (2013). Enhancing microvascular formation and vessel maturation through temporal control over multiple pro-angiogenic and pro-maturation factors. Biomaterials, 34(36), 9201-9209.
18. Rohlenova, K., Veys, K., Miranda-Santos, I., De Bock, K., & Carmeliet, P. (2018). Endothelial cell metabolism in health and disease. Trends in cell biology, 28(3), 224-236.
19. Paku, S., DezsÅ?, K., Bugyik, E., Tóvári, J., Tímár, J., Nagy, P.,... & Döme, B. (2011). A new mechanism for pillar formation during tumor-induced intussusceptive angiogenesis: inverse sprouting. The American journal of pathology, 179(3), 1573- 1585.
20. Betz, C., Lenard, A., Belting, H. G., & Affolter, M. (2016). Cell behaviors and dynamics during angiogenesis. Development, 143(13), 2249-2260.
21. Fantin, A., Vieira, J. M., Gestri, G., Denti, L., Schwarz, Q., Prykhozhij, S., ... & Ruhrberg, C. (2010). Tissue macrophages act as cellular chaperones for vascular anastomosis downstream of VEGF-mediated endothelial tip cell induction. Blood, The Journal of the American Society of Hematology, 116(5), 829-840.
22. Gianni-Barrera, R., Bartolomeo, M., Vollmar, B., Djonov, V., & Banfi, A. (2014). Split for the cure: VEGF, PDGF-BB and intussusception in therapeutic angiogenesis. Biochemical Society Transactions, 42(6), 1637-1642.
23. Sivaraj, K. K., & Adams, R. H. (2016). Blood vessel formationand function in bone. Development, 143(15), 2706-2715.
24. Li, H., & Chang, J. (2013). Stimulation of proangiogenesis by calcium silicate bioactive ceramic. Acta biomaterialia, 9(2), 5379-5389.
25. Gorustovich, A. A., Roether, J. A., & Boccaccini, A. R. (2010). Effect of bioactive glasses on angiogenesis: a review of in vitro and in vivo evidences. Tissue Engineering Part B: Reviews, 16(2), 199-207.
26. Carmeliet, P. (2005). Angiogenesis in life, disease and medicine. Nature, 438(7070), 932-936.
27. Davidson, S. M. (2019). FAM3A–A mitochondrial route tothe stimulation of angiogenesis?. EBioMedicine, 43, 3-4.
28. Chapanian, R., & Amsden, B. G. (2010). Combined and sequential delivery of bioactive VEGF165 and HGF from poly (trimethylene carbonate) based photo-cross-linked elastomers. Journal of controlled release, 143(1), 53-63.
29. Chen, R. R., Silva, E. A., Yuen, W. W., Brock, A. A., Fischbach, C., Lin, A. S., ... & Mooney, D. J. (2007). Integrated approach to designing growth factor delivery systems. The FASEB Journal, 21(14), 3896-3903.
30. Greenberg, J. I., Shields, D. J., Barillas, S. G., Acevedo, L. M., Murphy, E., Huang, J., ... & Cheresh, D. A. (2008). A role for VEGF as a negative regulator of pericyte function and vessel maturation. Nature, 456(7223), 809-813.
31. Fagiani, E., & Christofori, G. (2013). Angiopoietins inangiogenesis. Cancer letters, 328(1), 18-26.
32. Thurston, G., Suri, C., Smith, K., McClain, J., Sato, T. N., Yancopoulos, G. D., & McDonald, D. M. (1999). Leakage- resistant blood vessels in mice transgenically overexpressing angiopoietin-1. Science, 286(5449), 2511-2514.
33. Folkman, J. (2007). Angiogenesis: an organizing principle for drug discovery?. Nature reviews Drug discovery, 6(4), 273- 286.
34. Sakurai, T., & Kudo, M. (2011). Signaling pathways governingtumor angiogenesis. Oncology, 81(Suppl. 1), 24-29.
35. Payne, L. B., Tewari, B. P., Dunkenberger, L., Bond, S., Savelli, A., Darden, J., ... & Chappell, J. C. (2022). Pericyte progenitor coupling to the emerging endothelium during vasculogenesis via connexin 43. Arteriosclerosis, thrombosis, and vascular biology, 42(4), e96-e114.
36. Kruse, K., Lee, Q. S., Sun, Y., Klomp, J., Yang, X., Huang, F., ... & Komarova, Y. A. (2019). N-cadherin signaling via Trio assembles adherens junctions to restrict endothelial permeability. Journal of Cell Biology, 218(1), 299-316.
37. Moccia, F., Negri, S., Shekha, M., Faris, P., & Guerra,G. (2019). Endothelial Ca2+ signaling, angiogenesis and vasculogenesis: just what it takes to make a blood vessel. International journal of molecular sciences, 20(16), 3962.
38. Annex, B. H. (2013). Therapeutic angiogenesis for criticallimb ischaemia. Nature Reviews Cardiology, 10(7), 387-396.
39. Johnson, K. E., & Wilgus, T. A. (2014). Vascular endothelial growth factor and angiogenesis in the regulation of cutaneous wound repair. Advances in wound care, 3(10), 647-661.
40. Hoeben, A. N. N., Landuyt, B., Highley, M. S., Wildiers, H., Van Oosterom, A. T., & De Bruijn, E. A. (2004). Vascular endothelial growth factor and angiogenesis. Pharmacological reviews, 56(4), 549-580.
41. Braghirolli, D. I., Helfer, V. E., Chagastelles, P. C., Dalberto,T. P., Gamba, D., & Pranke, P. (2017). Electrospun scaffolds functionalized with heparin and vascular endothelial growth factor increase the proliferation of endothelial progenitor cells. Biomedical Materials, 12(2), 025003.
42. Yoo, S. Y., & Kwon, S. M. (2013). Angiogenesis and its therapeutic opportunities. Mediators of inflammation, 2013(1), 127170.
43. Maharaj, A. S., Saint-Geniez, M., Maldonado, A. E., & D'Amore, P. A. (2006). Vascular endothelial growth factor localization in the adult. The American journal of pathology,
168(2), 639-648.
44. Chung, A. S., Lee, J., & Ferrara, N. (2010). Targeting the tumour vasculature: insights from physiological angiogenesis. Nature reviews cancer, 10(7), 505-514.
45. Cadenas, S. (2018). ROS and redox signaling in myocardial ischemia-reperfusion injury and cardioprotection. Free Radical Biology and Medicine, 117, 76-89.
46. Kivelä, R., Bry, M., Robciuc, M. R., Räsänen, M., Taavitsainen, M., Silvola, J. M., ... & Alitalo, K. (2014). VEGFâ?Bâ?induced vascular growth leads to metabolic reprogramming and ischemia resistance in the heart. EMBO molecular medicine, 6(3), 307-321.
47. Groppa, E., Brkic, S., Bovo, E., Reginato, S., Sacchi, V., Di Maggio, N., ... & Banfi, A. (2015). VEGF dose regulates vascular stabilization through Semaphorin3A and the Neuropilinâ?1+ monocyte/TGFâ?β1 paracrine axis. EMBO molecular medicine, 7(10), 1366-1384.
48. Grunewald, M., Kumar, S., Sharife, H., Volinsky, E., Gileles- Hillel, A., Licht, T., ... & Keshet, E. (2021). Counteracting age-related VEGF signaling insufficiency promotes healthy aging and extends life span. Science, 373(6554), eabc8479.
49. Rissanen, T. T., Markkanen, J. E., Gruchala, M., Heikura, T., Puranen, A., Kettunen, M. I., ... & YlaÌ?-Herttuala, S. (2003). VEGF-D is the strongest angiogenic and lymphangiogenic effector among VEGFs delivered into skeletal muscle via adenoviruses. Circulation research, 92(10), 1098-1106.
50. Wu, M., Pokreisz, P., Swinnen, M., Caluwe, E., Gillijns, H., Vanden Driessche, N., ... & Janssens, S. (2017). Sustained placental growth factor-2 treatment does not aggravate advanced atherosclerosis in ischemic cardiomyopathy. Journal of Cardiovascular Translational Research, 10(4), 348-358.
51. Khurana, R., Moons, L., Shafi, S., Luttun, A., Collen, D., Martin, J. F., ... & Zachary, I. C. (2005). Placental growth factor promotes atherosclerotic intimal thickening and macrophage accumulation. Circulation, 111(21), 2828-2836.
52. Murasawa, S., Llevadot, J., Silver, M., Isner, J. M., Losordo,D. W., & Asahara, T. (2002). Constitutive human telomerase reverse transcriptase expression enhances regenerative properties of endothelial progenitor cells. circulation, 106(9), 1133-1139.
53. Goonoo, N., & Bhaw-Luximon, A. (2019). Mimicking growth factors: role of small molecule scaffold additives in promoting tissue regeneration and repair. RSC advances, 9(32), 18124- 18146.
54. Risau, W. (1990). Angiogenic growth factors. Progress in growth factor research, 2(1), 71-79.
55. Chung, J. C. Y., & Shum-Tim, D. (2012). Neovascularizationin tissue engineering. Cells, 1(4), 1246-1260.
56. Carmeliet, P., & Jain, R. K. (2011). Molecular mechanisms and clinical applications of angiogenesis. Nature, 473(7347), 298-307.
57. Zhao, W., Han, Q., Lin, H., Gao, Y., Sun, W., Zhao, Y., ... &Dai, J. (2008). Improved neovascularization and wound repair by targeting human basic fibroblast growth factor (bFGF) to fibrin. Journal of molecular medicine, 86, 1127-1138.
58. Li, J., Wei, Y., Liu, K., Yuan, C., Tang, Y., Quan, Q., ... &Yang, L. (2010). Synergistic effects of FGF-2 and PDGF-BB on angiogenesis and muscle regeneration in rabbit hindlimb ischemia model. Microvascular research, 80(1), 10-17.
59. Nikol, S., Baumgartner, I., Van Belle, E., Diehm, C., Visoná, A., Capogrossi, M. C., ... & Meyer, F. (2008). Therapeutic angiogenesis with intramuscular NV1FGF improves amputation-free survival in patients with critical limb ischemia. Molecular Therapy, 16(5), 972-978.
60. Cai, S., Liu, Y., Shu, X. Z., & Prestwich, G. D. (2005). Injectable glycosaminoglycan hydrogels for controlled release of human basic fibroblast growth factor. Biomaterials, 26(30), 6054-6067.
61. Oh, J. K., Drumright, R., Siegwart, D. J., & Matyjaszewski,K. (2008). The development of microgels/nanogels for drug delivery applications. Progress in polymer science, 33(4), 448-477.
62. Phan, V. G., Thambi, T., Duong, H. T. T., & Lee, D. S. (2016).Poly (amino carbonate urethane)-based biodegradable, temperature and pH-sensitive injectable hydrogels for sustained human growth hormone delivery. Scientific reports, 6(1), 29978.
63. Duan, H., Li, X., Wang, C., Hao, P., Song, W., Li, M., ... &Yang, Z. (2016). Functional hyaluronate collagen scaffolds induce NSCs differentiation into functional neurons in repairing the traumatic brain injury. Acta biomaterialia, 45, 182-195.
64. Xiang, Q., Xiao, J., Zhang, H., Zhang, X., Lu, M., Zhang, H.,... & Li, X. (2011). Preparation and characterisation of bFGF- encapsulated liposomes and evaluation of wound-healing activities in the rat. Burns, 37(5), 886-895.
65. Mukherjee, S., & Patra, C. R. (2016). Therapeutic application of anti-angiogenic nanomaterials in cancers. Nanoscale, 8(25), 12444-12470.
66. Powell, R. J., Goodney, P., Mendelsohn, F. O., Moen, E. K., Annex, B. H., & HGF-0205 Trial Investigators. (2010). Safety and efficacy of patient specific intramuscular injection of HGF plasmid gene therapy on limb perfusion and wound healing in patients with ischemic lower extremity ulceration: results of the HGF-0205 trial. Journal of vascular surgery, 52(6), 1525-1530.
67. Powell, R. J., Simons, M., Mendelsohn, F. O., Daniel, G., Henry, T. D., Koga, M., ... & Annex, B. H. (2008). Results of a double-blind, placebo-controlled study to assess the safety of intramuscular injection of hepatocyte growth factor plasmid to improve limb perfusion in patients with critical limb ischemia. Circulation, 118(1), 58-65.
68. Morishita, R., Shimamura, M., Takeya, Y., Nakagami, H., Chujo, M., Ishihama, T., ... & Rakugi, H. (2020). Combined analysis of clinical data on HGF gene therapy to treat critical limb ischemia in Japan. Current Gene Therapy, 20(1), 25-35.
69. Sanada, F., Fujikawa, T., Shibata, K., Taniyama, Y., Rakugi, H., & Morishita, R. (2020). Therapeutic angiogenesis using HGF plasmid. Annals of Vascular Diseases, 13(2), 109-115.
70. Pyun, W. B., Hahn, W., Kim, D. S., Yoo, W. S., Lee, S. D., Won, J. H., ... & Kim, S. (2010). Naked DNA expressing two isoforms of hepatocyte growth factor induces collateral artery augmentation in a rabbit model of limb ischemia. Gene therapy, 17(12), 1442-1452.
71. Lu, Q., Yao, Y., Yao, Y., Liu, S., Huang, Y., Lu, S., ... & Wang,Q. (2012). Angiogenic factor AGGF1 promotes therapeutic
angiogenesis in a mouse limb ischemia model.
72. Yao, Y., Li, Y., Song, Q., Hu, C., Xie, W., Xu, C., ... & Wang,Q. K. (2019). Angiogenic factor AGGF1-primed endothelial progenitor cells repair vascular defect in diabetic mice. Diabetes, 68(8), 1635-1648.
73. Wang, J., Peng, H., Timur, A. A., Pasupuleti, V., Yao, Y., Zhang, T., ... & Wang, Q. (2021). Receptor and molecular mechanism of AGGF1 signaling in endothelial cell functions and angiogenesis. Arteriosclerosis, thrombosis, and vascular biology, 41(11), 2756-2769.
74. Wu, J., Heemskerk, J. W., & Baaten, C. C. (2021). Platelet membrane receptor proteolysis: implications for platelet function. Frontiers in cardiovascular medicine, 7, 608391.
75. Heuberger, D. M., & Schuepbach, R. A. (2019). Protease- activated receptors (PARs): mechanisms of action and potential therapeutic modulators in PAR-driven inflammatory diseases. Thrombosis journal, 17(1), 4.
76. Posma, J. J. N., Posthuma, J. J., & Spronk, H. M. H. (2016). Coagulation and nonâ?coagulation effects of thrombin. Journal of thrombosis and haemostasis, 14(10), 1908-1916.
77. Posma, J. J., Grover, S. P., Hisada, Y., Owens III, A. P., Antoniak, S., Spronk, H. M., & Mackman, N. (2019). Roles of coagulation proteases and PARs (protease-activated receptors) in mouse models of inflammatory diseases. Arteriosclerosis, thrombosis, and vascular biology, 39(1), 13-24.
78. Burzynski, L. C., Humphry, M., Pyrillou, K., Wiggins, K. A., Chan, J. N., Figg, N., ... & Clarke, M. C. (2019). The coagulation and immune systems are directly linked through the activation of interleukin-1α by thrombin. Immunity, 50(4), 1033-1042.
79. Fang, X., Liao, R., Yu, Y., Li, J., Guo, Z., & Zhu, T. (2019).Thrombin Induces Secretion of Multiple Cytokines and Expression of Proteaseâ?Activated Receptors in Mouse Mast Cell Line. Mediators of Inflammation, 2019(1), 4952131.
80. Jaberi, N., Soleimani, A., Pashirzad, M., Abdeahad, H., Mohammadi, F., Khoshakhlagh, M., ... & Hassanian, S. M. (2019). Role of thrombin in the pathogenesis of atherosclerosis. Journal of Cellular Biochemistry, 120(4), 4757-4765.
81. Bea, F., Kreuzer, J., Preusch, M., Schaab, S., Isermann, B., Rosenfeld, M. E., ... & Blessing, E. (2006). Melagatran reduces advanced atherosclerotic lesion size and may promote plaque stability in Apolipoprotein E–deficient mice. Arteriosclerosis, thrombosis, and vascular biology, 26(12), 2787-2792.
82. Grebe, A., Hoss, F., & Latz, E. (2018). NLRP3 Inflammasome and the IL-1 Pathway in Atherosclerosis. Circulation research, 122(12), 1722-1740.
83. Latz, E., Xiao, T. S., & Stutz, A. (2013). Activation and regulation of the inflammasomes. Nature Reviews Immunology, 13(6), 397-411.
84. Galea, J., Armstrong, J., Gadsdon, P., Holden, H., Francis, S. E., & Holt, C. M. (1996). Interleukin-1β in coronary arteries of patients with ischemic heart disease. Arteriosclerosis, thrombosis, and vascular biology, 16(8), 1000-1006.
85. Joosten, L. A., Netea, M. G., Fantuzzi, G., Koenders, M. I., Helsen, M. M., Sparrer, H., ... & Van Den Berg, W. B. (2009). Inflammatory arthritis in caspase 1 gene–deficient mice: contribution of proteinase 3 to caspase 1–independent production of bioactive interleukinâ?1β. Arthritis & Rheumatism: Oficial Journal of the American College of Rheumatology, 60(12), 3651-3662.
86. Weber, A., Wasiliew, P., & Kracht, M. (2010). Interleukin-1(IL-1) pathway. Science signaling, 3(105), cm1-cm1.
87. Weber, A., Wasiliew, P., & Kracht, M. (2010). Interleukin-1(IL-1) pathway. Science signaling, 3(105), cm1-cm1.
88. Loppnow, H., & Libby, P. (1990). Proliferating or interleukin 1-activated human vascular smooth muscle cells secrete copious interleukin 6. The Journal of clinical investigation, 85(3), 731-738.
89. Dinarello, C. A. (2009). Immunological and inflammatory functions of the interleukin-1 family. Annual review of immunology, 27(1), 519-550.
90. Beltrami-Moreira, M., Vromman, A., Sukhova, G. K., Folco,
E. J., & Libby, P. (2016). Redundancy of IL-1 isoform signaling and its implications for arterial remodeling. PLoS One, 11(3), e0152474.
91. Libby, P. (2013). Collagenases and cracks in the plaque. The Journal of clinical investigation, 123(8), 3201-3203.
92. Libby, P. (2017). Interleukin-1 beta as a target for atherosclerosis therapy: biological basis of CANTOS and beyond. Journal of the American College of Cardiology, 70(18), 2278-2289.
93. Vromman, A., Ruvkun, V., Shvartz, E., Wojtkiewicz, G., Santos Masson, G., Tesmenitsky, Y., ... & Libby, P. (2019). Stage-dependent differential effects of interleukin-1 isoforms on experimental atherosclerosis. European heart journal, 40(30), 2482-2491.
94. Kamari, Y., Shaish, A., Shemesh, S., Vax, E., Grosskopf, I., Dotan, S., ... & Harats, D. (2011). Reduced atherosclerosis and inflammatory cytokines in apolipoprotein-E-deficient mice lacking bone marrow-derived interleukin-1α. Biochemical and biophysical research communications, 405(2), 197-203.
95. Libby, P., Warner, S. J. C., & Friedman, G. B. (1988). Interleukin 1: a mitogen for human vascular smooth muscle cells that induces the release of growth-inhibitory prostanoids. The Journal of clinical investigation, 81(2), 487-498.
96. Tsioufis, P., Theofilis, P., Tsioufis, K., & Tousoulis, D. (2022). The impact of cytokines in coronary atherosclerotic plaque: current therapeutic approaches. International Journal of Molecular Sciences, 23(24), 15937.
97. Dinarello, C. A. (2018). Overview of the ILâ?1 family in innate inflammation and acquired immunity. Immunological reviews, 281(1), 8-27.
98. Herder, C., De Las Heras Gala, T., Carstensen-Kirberg, M., Huth, C., Zierer, A., Wahl, S., ... & Thorand, B. (2017). Circulating levels of interleukin 1-receptor antagonist and risk of cardiovascular disease: meta-analysis of six population- based cohorts. Arteriosclerosis, thrombosis, and vascular biology, 37(6), 1222-1227.
99. Mai, W., & Liao, Y. (2020). Targeting IL-1β in the Treatment of Atherosclerosis. Frontiers in immunology, 11, 589654.
100. Lee, Y. W., & Hirani, A. A. (2006). Role of interleukin-4 in atherosclerosis. Archives of pharmacal research, 29, 1-15.
101. Galéa, P., Thibault, G., Lacord, M., Bardos, P., & Lebranchu, Y. (1993). IL-4, but not tumor necrosis factor-alpha, increases endothelial cell adhesiveness for lymphocytes by activating a cAMP-dependent pathway. Journal of immunology (Baltimore, Md.: 1950), 151(2), 588-596.
102. Lee, Y. W., Hennig, B., & Toborek, M. (2003). Redox-regulated mechanisms of IL-4-induced MCP-1 expression in human vascular endothelial cells. American Journal of Physiology- Heart and Circulatory Physiology, 284(1), H185-H192.
103. Iademarco, M. F., Barks, J. L., & Dean, D. C. (1995). Regulation of vascular cell adhesion molecule-1 expression by IL-4 and TNF-alpha in cultured endothelial cells. The Journal of clinical investigation, 95(1), 264-271.
104. Ali, M., Girgis, S., Hassan, A., Rudick, S., & Becker, R. C. (2018). Inflammation and coronary artery disease: from pathophysiology to Canakinumab Anti-Inflammatory Thrombosis Outcomes Study (CANTOS). Coronary Artery Disease, 29(5), 429-437.
105. Silveira, A., McLeod, O., Strawbridge, R. J., Gertow, K., Sennblad, B., Baldassarre, D., ... & Hamsten,A. (2015). Plasma IL-5 concentration and subclinical carotid atherosclerosis. Atherosclerosis, 239(1), 125-130.
106. Ishigami, T., Abe, K., Aoki, I., Minegishi, S., Ryo, A., Matsunaga, S., ... & Endo, Y. (2013). Antiâ?interleukinâ?5 and multiple autoantibodies are associated with human atherosclerotic diseases and serum interleukinâ?5 levels. The FASEB Journal, 27(9), 3437-3445.
107. Zhao, W., Lei, T., Li, H., Sun, D., Mo, X., Wang, Z., ... & Ou, H. (2015). Macrophage-specific overexpression of interleukin-5 attenuates atherosclerosis in LDL receptor-deficient mice. Gene therapy, 22(8), 645-652.
108. Ren, W., Wang, Z., Wang, J., Wu, Z., Ren, Q., Yu, A., & Ruan, Y. (2020). IL-5 overexpression attenuates aortic dissection by reducing inflammation and smooth muscle cell apoptosis. Life Sciences, 241, 117144.
109. Fard, T. K., Ahmadi, R., Akbari, T., Moradi, N., Fadaei, R., Fard, M. K., & Fallah, S. (2021). Klotho, FOXO1 and cytokines associations in patients with coronary artery disease. Cytokine, 141, 155443.
110. Schrader, J. W., Moyer, C., Ziltener, H. J., & Reinisch, C. L. (1991). Release of the cytokines colony-stimulating factor-1, granulocyte-macrophage colony-stimulating factor, and IL-6 by cloned murine vascular smooth muscle cells. Journal of immunology (Baltimore, Md.: 1950), 146(11), 3799-3808.
111. Reiss,A. B., Siegart, N. M., & De Leon, J. (2017). Interleukin-6 in atherosclerosis: atherogenic or atheroprotective?. Clinical Lipidology, 12(1), 14-23.
112. Schaper, F., & Rose-John, S. (2015). Interleukin-6: Biology, signaling and strategies of blockade. Cytokine & growthfactor reviews, 26(5), 475-487.
113. Xing, Z., Gauldie, J., Cox, G., Baumann, H., Jordana, M., Lei, X. F., & Achong, M. K. (1998). IL-6 is an antiinflammatory cytokine required for controlling local or systemic acute inflammatory responses. The Journal of clinical investigation, 101(2), 311-320.
114. Rose-John, S. (2012). IL-6 trans-signaling via the soluble IL-6 receptor: importance for the pro-inflammatory activities of IL-6. International journal of biological sciences, 8(9), 1237.
115. Arya, A. K., Tripathi, R., Kumar, S., & Tripathi, K. (2014). Recent advances on the association of apoptosis in chronic non healing diabetic wound. World journal of diabetes, 5(6), 756.
116. Li, R., Paul, A., Ko, K. W., Sheldon, M., Rich, B. E., Terashima, T., ... & Oka, K. (2012). Interleukin-7 induces recruitment of monocytes/macrophages to endothelium. European heart journal, 33(24), 3114-3123.
117. Morishita, R., Makino, H., Aoki, M., Hashiya, N., Yamasaki, K., Azuma, J., ... & Ogihara, T. (2011). Phase I/IIa clinical trial of therapeutic angiogenesis using hepatocyte growth factor gene transfer to treat critical limb ischemia. Arteriosclerosis, thrombosis, and vascular biology, 31(3), 713-720.
118. Zhang, W., Tang, T., Nie, D., Wen, S., Jia, C., Zhu, Z., ... & Cheng, X. (2015). IL-9 aggravates the development of atherosclerosis in ApoE−/− mice. Cardiovascular research, 106(3), 453-464.
119. von der Thüsen, J. H., Kuiper, J., Fekkes, M. L., de Vos, P., van Berkel, T. J., & Biessen, E. A. (2001). Attenuation of atherogenesis by systemic and local adenovirusâ?mediated gene transfer of interleukinâ?10 in LDLrâ?/â?mice. The FASEB Journal, 15(14), 1-19.
120. Mittal, S. K., Cho, K. J., Ishido, S., & Roche, P. A. (2015). Interleukin 10 (IL-10)-mediated immunosuppression: March-I induction regulates antigen presentation by macrophages but not dendritic cells. Journal of Biological Chemistry, 290(45), 27158-27167.
121. Han, X., & Boisvert, W. A. (2015). Interleukin-10 protects against atherosclerosis by modulating multiple atherogenic macrophage function. Thrombosis and haemostasis, 113(03), 505-512.
122. Tan, H., Dan, G., Gong, H., & Cao, L. (2005). Purification and characterization of recombinant truncated human interleukin-11 expressed as fusion protein in Escherichia coli. Biotechnology letters, 27, 905-910.
123. Demyanets, S., Huber, K., & Wojta, J. (2012). Vascular effects of glycoprotein130 ligands—Part I: Pathophysiological role. Vascular pharmacology, 56(1-2), 34-46.
124. Abu El-Asrar, A. M., Ahmad, A., Allegaert, E., Siddiquei, M. M., Gikandi, P. W., De Hertogh, G., & Opdenakker, G. (2020). Interleukin-11 overexpression and M2 macrophage density are associated with angiogenic activity in proliferative diabetic retinopathy. Ocular immunology and inflammation, 28(4), 575-588.
125. Roger, I., Estornut, C., Ballester, B., Milara, J., & Cortijo, J. (2019). Role of IL-11 in vascular function of pulmonaryibrosis patients.
126. Elshabrawy, H. A., Volin, M. V., Essani, A. B., Chen, Z., McInnes, I. B., Van Raemdonck, K., ... & Shahrara, S. (2018). IL-11 facilitates a novel connection between RA joint fibroblasts and endothelial cells. Angiogenesis, 21, 215-228.
127. Lamertz, L., Rummel, F., Polz, R., Baran, P., Hansen, S., Waetzig, G. H., ... & Scheller, J. (2018). Soluble gp130 prevents interleukin-6 and interleukin-11 cluster signaling but not intracellular autocrine responses. Science signaling, 11(550), eaar7388.
128. Guo, Y. T., Lu, Y. Y., Lu, X., He, S., Li, S. J., Shao, S., ... & Gao, P. J. (2021). Krüppelâ?Like Factor 15/Interleukin 11 Axisâ? Mediated Adventitial Remodeling Depends on Extracellular Signalâ?Regulated Kinases 1 and 2 Activation in Angiotensin II–Induced Hypertension. Journal of the American Heart Association, 10(16), e020554.
129. Mahboubi, K., Biedermann, B. C., Carroll, J. M., & Pober, J. S. (2000). IL-11 activates human endothelial cells to resist immune-mediated injury. The Journal of Immunology, 164(7), 3837-3846.
130. Lim, W. W., Corden, B., Ng, B., Vanezis, K., D’Agostino, G., Widjaja, A. A., ... & Cook, S. A. (2020). Interleukin-11 is important for vascular smooth muscle phenotypic switching and aortic inflammation, fibrosis and remodeling in mouse models. Scientific reports, 10(1), 17853.
131. Widjaja, A. A., Viswanathan, S., Jinrui, D., Singh, B. K., Tan, J., Wei Ting, J. G., ... & Cook, S. A. (2021). Molecular dissection of pro-fibrotic IL11 signaling in cardiac and pulmonary fibroblasts. Frontiers in Molecular Biosciences, 8, 740650.
132. Ye, J., Wang, Y., Wang, Z., Liu, L., Yang, Z., Wang, M., ... & Wan, J. (2020). Roles and mechanisms of interleukin-12 family members in cardiovascular diseases: opportunities and challenges. Frontiers in Pharmacology, 11, 129.
133. Andrews, C., McLean, M. H., & Durum, S. K. (2016). Interleukin-27 as a novel therapy for inflammatory bowel disease: a critical review of the literature. Inflammatory bowel diseases, 22(9), 2255-2264.
134. Kan, X., Wu, Y., Ma, Y., Zhang, C., Li, P., Wu, L., ... & Du, J. (2016). Deficiency of IL-12p35 improves cardiac repair after myocardial infarction by promoting angiogenesis. Cardiovascular research, 109(2), 249-259.
135. Fichtner-Feigl, S., Strober, W., Kawakami, K., Puri, R. K., & Kitani, A. (2006). IL-13 signaling through the IL-13α2 receptor is involved in induction of TGF-β1 production and fibrosis. Nature medicine, 12(1), 99-106.
136. Fatkhullina, A. R., Peshkova, I. O., & Koltsova, E. K. (2016). The role of cytokines in the development of atherosclerosis. Biochemistry (Moscow), 81, 1358-1370.
137. Cardiloâ?Reis, L., Gruber, S., Schreier, S. M., Drechsler, M., Papacâ?Milicevic, N., Weber, C., ... & Binder, C. J. (2012). Interleukinâ?13 protects from atherosclerosis and modulates plaque composition by skewing the macrophage phenotype. EMBO molecular medicine, 4(10), 1072-1086.
138. Rossol, M., Heine, H., Meusch, U., Quandt, D., Klein, C., Sweet, M. J., & Hauschildt, S. (2011). LPS-induced cytokine production in human monocytes and macrophages. Critical Reviews™ in Immunology, 31(5).
139. Guo, L., Liu, M. F., Huang, J. N., Li, J. M., Jiang, J., & Wang, J. A. (2020). Role of interleukinâ?15 in cardiovascular diseases. Journal of cellular and molecular medicine, 24(13), 7094-7101.
140. Bettelli, E., Korn, T., Oukka, M., & Kuchroo, V. K. (2008). Induction and effector functions of TH17 cells. Nature, 453(7198), 1051-1057.
141. Fossiez, F., Djossou, O., Chomarat, P., Flores-Romo, L., Ait-Yahia, S., Maat, C., ... & Lebecque, S. (1996). T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. Journal of Experimental Medicine, 183(6), 2593-2603.
142. McGeachy, M. J., Cua, D. J., & Gaffen, S. L. (2019). The IL- 17 family of cytokines in health and disease. Immunity, 50(4), 892-906.
143. Faour, W. H., Mancini, A., He, Q. W., & Di Battista, J. A. (2003). T-cell-derived interleukin-17 regulates the level and stability of cyclooxygenase-2 (COX-2) mRNA through restricted activation of the p38 mitogen-activated protein kinase cascade: role of distal sequences in the 3′-untranslated region of COX-2 mRNA. Journal of Biological Chemistry, 278(29), 26897-26907.
144. Kidani, Y., & Bensinger, S. J. (2017). Reviewing the impact of lipid synthetic flux on Th17 function. Current opinion in immunology, 46, 121-126.
145. Taleb, S., Tedgui, A., & Mallat, Z. (2010). Interleukin-17: friend or foe in atherosclerosis?. Current opinion in lipidology, 21(5), 404-408.
146. O'Connor Jr, W., Kamanaka, M., Booth, C. J., Town, T., Nakae, S., Iwakura, Y., ... & Flavell, R. A. (2009). A protective function for interleukin 17A in T cell–mediated intestinal inflammation. Nature immunology, 10(6), 603-609.
147. Taleb, S., Romain, M., Ramkhelawon, B., Uyttenhove, C., Pasterkamp, G., Herbin, O., ... & Mallat, Z. (2009). Loss of SOCS3 expression in T cells reveals a regulatory role for interleukin-17 in atherosclerosis. Journal of Experimental Medicine, 206(10), 2067-2077.
148. Danzaki, K., Matsui, Y., Ikesue, M., Ohta, D., Ito, K., Kanayama, M., ... & Uede, T. (2012). Interleukin-17A deficiency accelerates unstable atherosclerotic plaque formation in apolipoprotein E-deficient mice. Arteriosclerosis, thrombosis, and vascular biology, 32(2), 273-280.
149. Erbel, C., Dengler, T. J., Wangler, S., Lasitschka, F., Bea, F., Wambsganss, N., ... & Gleissner, C. A. (2011). Expression of IL-17A in human atherosclerotic lesions is associated with increased inflammation and plaque vulnerability. Basic research in cardiology, 106, 125-134.
150. Eid, R. E., Rao, D. A., Zhou, J., Lo, S. F. L., Ranjbaran, H., Gallo, A., ... & Tellides, G. (2009). Interleukin-17 and interferon-γ are produced concomitantly by human coronary artery–infiltrating T cells and act synergistically on vascular smooth muscle cells. Circulation, 119(10), 1424-1432.
151. Gerdes, N., Sukhova, G. K., Libby, P., Reynolds, R. S., Young, . L., & Schönbeck, U. (2002). Expression of interleukin (IL)-18 and functional IL-18 receptor on human vascular endothelial cells, smooth muscle cells, and macrophages: implications for atherogenesis. The Journal of experimental medicine, 195(2), 245-257.
152. Netea, M. G., Kullberg, B. J., Verschueren, I., & Meer, J. W. V. D. (2000). Interleukinâ?18 induces production of proinflammatory cytokines in mice: no intermediate role for the cytokines of the tumor necrosis factor family and interleukinâ?1β. European journal of immunology, 30(10), 3057-3060.
153. Kannan, Y., Yu, J., Raices, R. M., Seshadri, S., Wei, M., Caligiuri, M. A., & Wewers, M. D. (2011). IκBζ augments IL- 12–and IL-18–mediated IFN-γ production in human NK cells. Blood, The Journal of the American Society of Hematology, 117(10), 2855-2863.
154. Gallagher, G. (2010). Interleukin-19: multiple roles in immune regulation and disease. Cytokine & growth factor reviews, 21(5), 345-352.
155. Gabunia, K., Ellison, S., Kelemen, S., Kako, F., Cornwell, W. D., Rogers, T. J., ... & Autieri, M. V. (2016). IL-19 halts progression of atherosclerotic plaque, polarizes, and increases cholesterol uptake and efflux in macrophages. The American journal of pathology, 186(5), 1361-1374.
156. Khallou-Laschet, J., Varthaman, A., Fornasa, G., Compain, C., Gaston, A. T., Clement, M., ... & Caligiuri, G. (2010). Macrophage plasticity in experimental atherosclerosis. PloS one, 5(1), e8852.
157. Williams, K. J., Feig, J. E., & Fisher, E. A. (2007). Cellular and molecular mechanisms for rapid regression of atherosclerosis: from bench top to potentially achievable clinical goal. Current opinion in lipidology, 18(4), 443-450.
158. Francis, A. A., & Pierce, G. N. (2011). An integrated approach for the mechanisms responsible for atherosclerotic plaque regression. Experimental & Clinical Cardiology, 16(3), 77.
159. Moore, K. J., & Freeman, M. W. (2006). Scavenger receptors in atherosclerosis: beyond lipid uptake. Arteriosclerosis, thrombosis, and vascular biology, 26(8), 1702-1711.
160. Rubic, T., & Lorenz, R. L. (2006). Downregulated CD36 and oxLDL uptake and stimulated ABCA1/G1 and cholesterol efflux as anti-atherosclerotic mechanisms of interleukin-10. Cardiovascular research, 69(2), 527-535.
161. Moore, K. J., Kunjathoor, V. V., Koehn, S. L., Manning, J. J., Tseng, A. A., Silver, J. M., ... & Freeman, M. W. (2005). Loss of receptor-mediated lipid uptake via scavenger receptor A or CD36 pathways does not ameliorate atherosclerosis in hyperlipidemic mice. The Journal of clinical investigation, 115(8), 2192-2201.
162. Kozarsky, K. F., Donahee, M. H., Glick, J. M., Krieger, M., & Rader, D. J. (2000). Gene transfer and hepatic overexpression of the HDL receptor SR-BI reduces atherosclerosis in the cholesterol-fed LDL receptor–deficient mouse. Arteriosclerosis, thrombosis, and vascular biology, 20(3), 721-727.
163. Mantovani, A., Sozzani, S., Locati, M., Allavena, P., & Sica, A. (2002). Macrophage polarization: tumor-associated crophages as a paradigm for polarized M2 mononuclear phagocytes. Trends in immunology, 23(11), 549-555.
164. Ellison, S., Gabunia, K., Kelemen, S. E., England, R. N., Scalia, R., Richards, J. M., ... & Autieri, M. V. (2013). Attenuation of experimental atherosclerosis by interleukin-19. Arteriosclerosis, thrombosis, and vascular biology, 33(10), 2316-2324.
165. Kunz, S., Wolk, K., Witte, E., Witte, K., Doecke, W. D., Volk, H. D., ... & Sabat, R. (2006). Interleukin (IL)â?19, ILâ?20 and ILâ?24 are produced by and act on keratinocytes and are distinct from classical ILs. Experimental dermatology, 15(12), 991- 1004.
166. Oral, H. B., Kotenko, S. V., Yılmaz, M., Mani, O., Zumkehr, J., Blaser, K., ... & Akdis, M. (2006). Regulation of T cells and cytokines by the interleukinâ?10 (ILâ?10)â?family cytokines ILâ?19, ILâ?20, ILâ?22, ILâ?24 andILâ?26. European journal of immunology, 36(2), 380-388.
167. Commins, S., Steinke, J. W., & Borish, L. (2008). The extended il-10 superfamily: Il-10, il-19, il-20, il-22, il-24, il-26, il- 28, and il-29. Journal of Allergy and Clinical Immunology, 121(5), 1108-1111.
168. Chen, W. Y., Cheng, B. C., Jiang, M. J., Hsieh, M. Y., & Chang, M. S. (2006). IL-20 Is expressed in atherosclerosis plaques and promotes atherosclerosis in apolipoprotein E– deficient mice. Arteriosclerosis, thrombosis, and vascular biology, 26(9), 2090-2095.
169. Hsieh, M. Y., Chen, W. Y., Jiang, M. J., Cheng, B. C., Huang,
T. Y., & Chang, M. S. (2006). Interleukin-20 promotes angiogenesis in a direct and indirect manner. Genes & Immunity, 7(3), 234-242.
170. Xia, Q., Xiang, X., Patel, S., Puranik, R., Xie, Q., & Bao, S. (2012). Characterisation of IL-22 and interferon-gamma- inducible chemokines in human carotid plaque. International journal of cardiology, 154(2), 187-189.
171. Rattik, S., Hultman, K., Rauch, U., Söderberg, I., Sundius, L., Ljungcrantz, I., ... & Nilsson, J. (2015). IL-22 affects smooth muscle cell phenotype and plaque formation in apolipoprotein E knockout mice. Atherosclerosis, 242(2), 506-514.
172. Chellan, B., Yan, L., Sontag, T. J., Reardon, C. A., & Bow- man, M. A. H. (2014). IL-22 is induced by S100/calgranulin and impairs cholesterol efflux in macrophages by downregu- lating ABCG1. Journal of lipid research, 55(3), 443-454.
173. Che, Y., Su, Z., & Xia, L. (2020). Effects of IL-22 on cardi- ovascular diseases. International Immunopharmacology, 81, 106277.
174. Luo, J. W., Hu, Y., Liu, J., Yang, H., & Huang, P. (2021). In- terleukin-22: a potential therapeutic target in atherosclerosis.
Molecular medicine, 27, 1-13.
175. Korn, T., Bettelli, E., Oukka, M., & Kuchroo, V. K. (2009). IL-17 and Th17 Cells. Annual review of immunology, 27(1), 485-517.
176. Wang, J., Zhao, P., Gao, Y., Zhang, F., Yuan, X., Jiao, Y., & Gong, K. (2019). The effects of anti-IL-23p19 therapy on ath- erosclerosis development in ApoE−/− mice. Journal of Inter- feron & Cytokine Research, 39(9), 564-571.
177. Abbas, A., Gregersen, I., Holm, S., Daissormont, I., Bjerkeli, V., Krohg-Sørensen, K., ... & Skjelland, M. (2015). Interleu- kin 23 levels are increased in carotid atherosclerosis: possible role for the interleukin 23/interleukin 17 axis. Stroke, 46(3), 793-799.
178. Subramanian, M., Thorp, E., & Tabas, I. (2015). Identification of a non-growth factor role for GM-CSF in advanced athero- sclerosis: promotion of macrophage apoptosis and plaque ne- crosis through IL-23 signaling. Circulation research, 116(2), e13-e24.
179. Fatkhullina, A. R., Peshkova, I. O., Dzutsev, A., Aghayev, T., McCulloch, J. A., Thovarai, V., ... & Koltsova, E. K. (2018). An interleukin-23-interleukin-22 axis regulates intestinal mi- crobial homeostasis to protect from diet-induced atherosclero- sis. Immunity, 49(5), 943-957.
180. Engelbertsen, D., Depuydt, M. A., Verwilligen, R. A., Rattik, S., Levinsohn, E., Edsfeldt, A., ... & Lichtman, A. H. (2018). IL 23R Deficiency Does Not Impact Atherosclerotic Plaque Development in Mice. Journal of the American Heart Associ- ation, 7(8), e008257.
181. Vargas-Alarcón, G., Posadas-Romero, C., Villarreal-Molina, T., Alvarez-León, E., Angeles-Martinez, J., Posadas-Sanchez, R., ... & Kimura-Hayama, E. (2014). IL-24 gene polymor- phisms are associated with cardiometabolic parameters and cardiovascular risk factors but not with premature coronary artery disease: the genetics of atherosclerotic disease Mexi- can study. Journal of Interferon & Cytokine Research, 34(9), 659-666.
182. Fort, M. M., Cheung, J., Yen, D., Li, J., Zurawski, S. M., Lo, S., ... & Rennick, D. M. (2001). IL-25 induces IL-4, IL-5,
and IL-13 and Th2-associated pathologies in vivo. Immunity, 15(6), 985-995.
183. Mantani, P. T., Dunér, P., Bengtsson, E., Ljungcrantz, I., Sun- dius, L., To, F., ... & Fredrikson, G. N. (2018). Interleukin-25 (IL-25) has a protective role in atherosclerosis development in the aortic arch in mice. Journal of Biological Chemistry, 293(18), 6791-6801.
184. Daugherty, A. (2002). Mouse models of atherosclerosis. The American journal of the medical sciences, 323(1), 3-10.
185. Mantani, P. T., Dunér, P., Bengtsson, E., Alm, R., Ljungcrantz, I., Söderberg, I., ... & Fredrikson, G. N. (2015). IL-25 inhib- its atherosclerosis development in apolipoprotein E deficient mice. PloS one, 10(1), e0117255.
186. Yoshida, H., & Hunter, C. A. (2015). The immunobiology of interleukin-27. Annual review of immunology, 33(1), 417-443.
187. Koltsova, E. K., Kim, G., Lloyd, K. M., Saris, C. J., Von Viet- inghoff, S., Kronenberg, M., & Ley, K. (2012). Interleukin-27 receptor limits atherosclerosis in Ldlr−/− mice. Circulation research, 111(10), 1274-1285.
188. Hirase, T., Hara, H., Miyazaki, Y., Ide, N., Nishimoto-Hazu- ku, A., Fujimoto, H., ... & Node, K. (2013). Interleukin 27 in- hibits atherosclerosis via immunoregulation of macrophages in mice. American Journal of Physiology-Heart and Circula- tory Physiology, 305(3), H420-H429.
189. Shoda, H., Fujio, K., Yamaguchi, Y., Okamoto, A., Sawada, T., Kochi, Y., & Yamamoto, K. (2006). Interactions between IL-32 and tumor necrosis factor alpha contribute to the exac-erbation of immune-inflammatory diseases. Arthritis research & therapy, 8, 1-13.
190. Park, M. H., Song, M. J., Cho, M. C., Moon, D. C., Yoon, D.Y., Han, S. B., & Hong, J. T. (2012). Interleukin 32 enhances cytotoxic effect of natural killer cells to cancer cells via acti- vation of death receptor 3. Immunology, 135(1), 63-72.
191. Zheng, C., Zheng, L., Yoo, J. K., Guo, H., Zhang, Y., Guo, X., ... & Zhang, Z. (2017). Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell, 169(7), 1342-1356.
192. Nold-Petry, C. A., Nold, M. F., Zepp, J. A., Kim, S. H., Voelkel, N. F., & Dinarello, C. A. (2009). IL-32–dependent effects of IL-1β on endothelial cell functions. Proceedings of the National Academy of Sciences, 106(10), 3883-3888.
193. Kobayashi, H., & Lin, P. C. (2009). Molecular characteriza- tion of IL-32 in human endothelial cells. Cytokine, 46(3), 351- 358.
194. Hong, J. T., Son, D. J., Lee, C. K., Yoon, D. Y., Lee, D. H., & Park, M. H. (2017). Interleukin 32, inflammation and cancer.
Pharmacology & therapeutics, 174, 127-137.
195. Zaidan, S. M., Leyre, L., Bunet, R., Larouche-Anctil, E., Tur- cotte, I., Sylla, M., ... & El-Far, M. (2019). Upregulation of IL-32 isoforms in virologically suppressed HIV-infected in- dividuals: potential role in persistent inflammation and tran- scription from stable HIV-1 reservoirs. JAIDS Journal of Ac- quired Immune Deficiency Syndromes, 82(5), 503-513.
196. Mohammad-Rezaei, M., Ahmadi, R., Rafiei, A., Khaledifar, A., Fattahi, S., Samiei-Sefat, A., ... & Bagheri, N. (2021). Se- rum levels of IL-32 in patients with coronary artery disease and its relationship with the serum levels of IL-6 and TNF-α. Molecular Biology Reports, 48(5), 4263-4271.
197. Yang, Z., Shi, L., Xue, Y., Zeng, T., Shi, Y., Lin, Y., & Liu, L. (2019). Interleukin-32 increases in coronary arteries and plasma from patients with coronary artery disease. Clinica chimica acta, 497, 104-109.
198. Heinhuis, B., Popa, C. D., van Tits, B. L., Kim, S. H., Zeeu- wen, P. L., van den Berg, W. B., ... & Joosten, L. A. (2013). Towards a role of interleukin-32 in atherosclerosis. Cytokine, 64(1), 433-440.
199. Kim, S. H., Han, S. Y., Azam, T., Yoon, D. Y., & Dinarello, C. A. (2005). Interleukin-32: a cytokine and inducer of TNFα.
Immunity, 22(1), 131-142.
200. Netea, M. G., Azam, T., Ferwerda, G., Girardin, S. E., Walsh, M., Park, J. S., ... & Kim, S. H. (2005). IL-32 synergizes with nucleotide oligomerization domain (NOD) 1 and NOD2 lig- ands for IL-1β and IL-6 production through a caspase 1-de- pendent mechanism. Proceedings of the National Academy of Sciences, 102(45), 16309-16314.
201. Choi, Y. S., Choi, H. J., Min, J. K., Pyun, B. J., Maeng, Y. S., Park, H., ... & Kwon, Y. G. (2009). Interleukin-33 in- duces angiogenesis and vascular permeability through ST2/ TRAF6-mediated endothelial nitric oxide production. Blood, The Journal of the American Society of Hematology, 114(14), 3117-3126.
202. Lu, J., Kang, J., Zhang, C., & Zhang, X. (2015). The role of L-33/ST2L signals in the immune cells. Immunology letters, 164(1), 11-17.
203. McLaren, J. E., Michael, D. R., Salter, R. C., Ashlin, T. G., Calder, C. J., Miller, A. M., ... & Ramji, D. P. (2010). IL-33 reduces macrophage foam cell formation. The Journal of Im- munology, 185(2), 1222-1229.
204. Zhang, H. F., Wu, M. X., Lin, Y. Q., Xie, S. L., Huang, T. C., Liu, P. M., ... & Wang, J. F. (2017). IL-33 promotes IL-10 pro- duction in macrophages: a role for IL-33 in macrophage foam cell formation. Experimental & molecular medicine, 49(11), e388-e388.
205. Miller, A. M., Xu, D., Asquith, D. L., Denby, L., Li, Y., Sattar, N., ... & Liew, F. Y. (2008). IL-33 reduces the development of atherosclerosis. Journal of Experimental Medicine, 205(2), 339-346.
206. Wei, X., Zhang, J., Gu, Q., Huang, M., Zhang, W., Guo, J., & Zhou, X. (2017). Reciprocal expression of IL-35 and IL-10 defines two distinct effector Treg subsets that are required for maintenance of immune tolerance. Cell reports, 21(7), 1853- 1869.
207. Sha, X., Meng, S., Li, X., Xi, H., Maddaloni, M., Pascual, D. W., ... & Yang, X. F. (2015). Interleukin-35 inhibits endotheli- al cell activation by suppressing MAPK-AP-1 pathway. Jour- nal of Biological Chemistry, 290(31), 19307-19318.
208. Urtz, N., Gaertner, F., von Bruehl, M. L., Chandraratne, S., Rahimi, F., Zhang, L., ... & Massberg, S. (2015). Sphingosine 1-phosphate produced by sphingosine kinase 2 intrinsically controls platelet aggregation in vitro and in vivo. Circulation research, 117(4), 376-387.
209. Liu, J., Lin, J., He, S., Wu, C., Wang, B., Liu, J., ... & Li, D.
(2018). Transgenic overexpression of IL-37 protects against atherosclerosis and strengthens plaque stability. Cellular Physiology and Biochemistry, 45(3), 1034-1050.
210. Sharma, S., Kulk, N., Nold, M. F., Gra f, R., Kim, S. H., Re- inhardt, D., ... & Bufler, P. (2008). The IL-1 family member 7b translocates to the nucleus and down-regulates proinflam- matory cytokines. The Journal of Immunology, 180(8), 5477- 5482.
211. Bulau, A. M., Nold, M. F., Li, S., Nold-Petry, C. A., Fink, M., Mansell, A., ... & Bufler, P. (2014). Role of caspase-1 in nuclear translocation of IL-37, release of the cytokine, and IL- 37 inhibition of innate immune responses. Proceedings of the National Academy of Sciences, 111(7), 2650-2655.
212. Nold, M. F., Nold-Petry, C. A., Zepp, J. A., Palmer, B. E., Bufler, P., & Dinarello, C. A. (2010). IL-37 is a fundamen- tal inhibitor of innate immunity. Nature immunology, 11(11), 1014-1022.
213. Chai, M., Ji, Q., Zhang, H., Zhou, Y., Yang, Q., Zhou, Y., ... & Zhao, Y. (2015). The protective effect of interleukin-37 on vascular calcification and atherosclerosis in apolipoprotein E-deficient mice with diabetes. Journal of Interferon & Cy- tokine Research, 35(7), 530-539.
214. Sanjabi, S., Zenewicz, L. A., Kamanaka, M., & Flavell, R. A. (2009). Anti-inflammatory and pro-inflammatory roles of TGF-β, IL-10, and IL-22 in immunity and autoimmunity. Current opinion in pharmacology, 9(4), 447-453.
215. Flusberg, D. A., & Sorger, P. K. (2015). Surviving apopto- sis: life–death signaling in single cells. Trends in cell biology, 25(8), 446-458.
216. Nash, M., McGrath, J. P., Cartland, S. P., Patel, S., & Ka- vurma, M. M. (2019). Tumour necrosis factor superfamily members in ischaemic vascular diseases. Cardiovascular Re- search, 115(4), 713-720.
217. Mackesy, D. Z., & Goalstone, M. L. (2014). Extracellular signal regulated kinase 5: Novel mediator of insulin and tu- mor necrosis factor α stimulated vascular cell adhesion mol- ecule 1 expression in vascular cells ç»?è??å¤?ä¿¡å·è°?è??æ¿?é?¶â?5:ä¸?ç§æ?°ç??è°?è??è?°å²?ç´ ä¸?è?¿ç?¤åæ»å? å α å?ºæ¿?ä¸?ç??è¡?管ç»?è??ä¸?ç??è¡?管ç»?è??ç²?é??å??åâ?1 表达ç??ä»?è´¨. Journal of Dia- betes, 6(6), 595-602.
218. Clinton, S. K., Underwood, R., Hayes, L., Sherman, M. L., Kufe, D. W., & Libby, P. (1992). Macrophage colony-stimu- lating factor gene expression in vascular cells and in experi- mental and human atherosclerosis. The American journal of pathology, 140(2), 301.
219. Tedgui, A., & Mallat, Z. (2006). Cytokines in atherosclerosis: pathogenic and regulatory pathways. Physiological reviews, 86(2), 515-581.
220. Ohta, H., Wada, H., Niwa, T., Kirii, H., Iwamoto, N., Fujii, H., ... & Seishima, M. (2005). Disruption of tumor necrosis factor-α gene diminishes the development of atherosclerosis in ApoE-deficient mice. Atherosclerosis, 180(1), 11-17.
221. Hashizume, M., & Mihara, M. (2012). Atherogenic effects of TNF-α and IL-6 via up-regulation of scavenger receptors. Cy- tokine, 58(3), 424-430.
222. Levy, Z., Rachmani, R., Trestman, S., Dvir, A., Shaish, A., Ravid, M., & Harats, D. (2003). Low-dose interferon-α ac- celerates atherosclerosis in an LDL receptor-deficient mouse model. European journal of internal medicine, 14(8), 479- 483.
223. Pepper, M. S., Mandriota, S. J., Vassalli, J. D., Orci, L., & Montesano, R. (1996). Angiogenesis-regulating cytokines: activities and interactions (pp. 31-67). Springer Berlin Hei- delberg.
224. Pammer, J., Reinisch, C., Birner, P., Pogoda, K., Sturzl, M., & Tschachler, E. (2006). Interferon-α prevents apoptosis of endothelial cells after short-term exposure but induces rep- licative senescence after continuous stimulation. Laboratory investigation, 86(10), 997-1007.
225. Ruszczak, Z., Detmar, M., Imcke, E., & Orfanos, C. E. (1990). Effects of rIFN alpha, beta, and gamma on the morphology, proliferation, and cell surface antigen expression of human dermal microvascular endothelial cells in vitro. Journal of in- vestigative dermatology, 95(6).
226. Minischetti, M., Vacca, A., Ribatti, D., Iurlaro, M., Ria, R., Pellegrino, A., ... & Dammacco, A. F. (2000). TNPâ?470 and recombinant human interferonâ?α2a inhibit angiogenesis syn- ergistically. British journal of haematology, 109(4), 829-837.
227. Cozzolino, F., Torcia, M., Lucibello, M., Morbidelli, L., Ziche, M., Platt, J., ... & Stern, D. (1993). Interferon-alpha and interleukin 2 synergistically enhance basic fibroblast growth factor synthesis and induce release, promoting en- dothelial cell growth. The Journal of clinical investigation, 91(6), 2504-2512.
228. Gomez, D., & Reich, N. C. (2003). Stimulation of primary human endothelial cell proliferation by IFN. The Journal of Immunology, 170(11), 5373-5381.
229. Sgonc, R., Fuerhapter, C., Boeck, G., Swerlick, R., Fritsch, P., & Sepp, N. (1998). Induction of apoptosis in human dermal microvascular endothelial cells and infantile hemangiomas by interferon-alpha. International archives of allergy and immu- nology, 117(3), 209-214.
230. Boshuizen, M. C., & de Winther, M. P. (2015). Interferons as essential modulators of atherosclerosis. Arteriosclerosis, thrombosis, and vascular biology, 35(7), 1579-1588.
231. Buttice, G., Miller, J., Wang, L., & Smith, B. D. (2006). In- terferon-γ induces major histocompatibility class II transacti- vator (CIITA), which mediates collagen repression and major histocompatibility class II activation by human aortic smooth muscle cells. Circulation research, 98(4), 472-479.
232. Ahmad, U., Ali, R., Lebastchi, A. H., Qin, L., Lo, S. F. L., Yakimov, A. O., ... & Tellides, G. (2010). IFN-γ Primes In- tact Human Coronary Arteries and Cultured Coronary Smooth Muscle Cells to Double-Stranded RNA-and Self-RNA–In- duced Inflammatory Responses by Upregulating TLR3 and Melanoma Differentiation-Associated Gene 5. The Journal of Immunology, 185(2), 1283-1294.
233. Zhou, Q. D., Chi, X., Lee, M. S., Hsieh, W. Y., Mkrtchyan, J. J., Feng, A. C., ... & Bensinger, S. J. (2020). Interferon-medi- ated reprogramming of membrane cholesterol to evade bacte- rial toxins. Nature immunology, 21(7), 746-755.
234. Mehta, N. N., Teague, H. L., Swindell, W. R., Baumer, Y., Ward, N. L., Xing, X., ... & Gudjonsson, J. E. (2017). IFN-γ and TNF-α synergism may provide a link between psoria- sis and inflammatory atherogenesis. Scientific reports, 7(1), 13831.
235. Gupta, S., Pablo, A. M., c Jiang, X., Wang, N., Tall, A. R., & Schindler, C. (1997). IFN-gamma potentiates atherosclerosis in ApoE knock-out mice. The Journal of clinical investiga- tion, 99(11), 2752-2761.
236. Serralheiro, P., Soares, A., Costa Almeida, C. M., & Verde, I. (2017). TGF-β1 in vascular wall pathology: Unraveling chronic venous insufficiency pathophysiology. International journal of molecular sciences, 18(12), 2534.
237. Chen, P. Y., Qin, L., Tellides, G., & Simons, M. (2014). Fi- broblast growth factor receptor 1 is a key inhibitor of TGFβ signaling in the endothelium. Science signaling, 7(344), ra90- ra90.
238. Ligi, D., Croce, L., Mosti, G., Raffetto, J. D., & Mannello, F. (2017). Chronic venous insufficiency: transforming growth factor-β isoforms and soluble endoglin concentration in differ- ent states of wound healing. International Journal of Molecu- lar Sciences, 18(10), 2206.
239.V.I. Golovina, E.I. Seliverstov, O.I. Efremova, I.A. Zolotukh- in, 15 (2021). Cytokines in pathogenesis of varicose veins, Flebologiya.
240. Tousoulis, D., Oikonomou, E., Economou, E. K., Crea, F., & Kaski, J. C. (2016). Inflammatory cytokines in atherosclero- sis: current therapeutic approaches. European heart journal, 37(22), 1723-1732.
241. Ramji, D. P., & Davies, T. S. (2015). Cytokines in athero- sclerosis: Key players in all stages of disease and promising therapeutic targets. Cytokine & growth factor reviews, 26(6), 673-685.
242. Adela, R., & Banerjee, S. K. (2015). GDFâ?15 as a target and biomarker for diabetes and cardiovascular diseases: a trans- lational prospective. Journal of diabetes research, 2015(1), 490842.
243. Wischhusen, J., Melero, I., & Fridman, W. H. (2020). Growth/ differentiation factor-15 (GDF-15): from biomarker to novel targetable immune checkpoint. Frontiers in immunology, 11, 951.
244. Wang, F. F., Chen, B. X., Yu, H. Y., Mi, L., Li, Z. J., & Gao, W. (2016). Correlation between growth differentiation factor-15 and collagen metabolism indicators in patients with myocar- dial infarction and heart failure. Journal of Geriatric Cardiol- ogy: JGC, 13(1), 88.
245. Wang, J., Wei, L., Yang, X., & Zhong, J. (2019). Roles of growth differentiation factor 15 in atherosclerosis and coro- nary artery disease. Journal of the American Heart Associa- tion, 8(17), e012826.
246. Lin, Y. D., Luo, C. Y., Hu, Y. N., Yeh, M. L., Hsueh, Y. C., Chang, M. Y., ... & Hsieh, P. C. (2012). Instructive nanofiber scaffolds with VEGF create a microenvironment for arteri- ogenesis and cardiac repair. Science translational medicine, 4(146), 146ra109-146ra109.
247. Bergsten, E., Uutela, M., Li, X., Pietras, K., Östman, A., Hel- din, C. H., ... & Eriksson, U. (2001). PDGF-D is a specific, protease-activated ligand for the PDGF β-receptor. Nature cell biology, 3(5), 512-516.
248. Cao, R., Bråkenhielm, E., Li, X., Pietras, K., Widenfalk, J., Östman, A., ... & Cao, Y. (2002). Angiogenesis stimulated by PDGFâ?CC, a novel member in the PDGF family, involves ac- tivation of PDGFRâ?aa andâ?ap receptors. The FASEB journal, 16(12), 1575-1583.
249. Li, X., Pontén, A., Aase, K., Karlsson, L., Abramsson, A., Uutela, M., ... & Eriksson, U. (2000). PDGF-C is a new pro- tease-activated ligand for the PDGF α-receptor. Nature cell biology, 2(5), 302-309.
250. Xue, Y., Lim, S., Yang, Y., Wang, Z., Jensen, L. D. E., Hed- lund, E. M., ... & Cao, Y. (2012). PDGF-BB modulates he- matopoiesis and tumor angiogenesis by inducing erythro- poietin production in stromal cells. Nature medicine, 18(1), 100-110.
251. Abramsson, A., Lindblom, P., & Betsholtz, C. (2003). En- dothelial and nonendothelial sources of PDGF-B regulate pericyte recruitment and influence vascular pattern formation in tumors. The Journal of clinical investigation, 112(8), 1142- 1151.
252. Hellberg, C., Östman, A., & Heldin, C. H. (2009). PDGF and
vessel maturation. Angiogenesis inhibition, 103-114.
253. Heldin, C. H., Eriksson, U., & Östman, A. (2002). New members of the platelet-derived growth factor family of mitogens.
Archives of biochemistry and biophysics, 398(2), 284-290.
254. Heldin, C. H., & Westermark, B. (1990). Commentary Signal transduction by the receptors for platelet-derived growth fac- tor. Journal of cell science, 96(2), 193-196.
255. Martino, M. M., Brkic, S., Bovo, E., Burger, M., Schaefer, D. J., Wolff, T., ... & Banfi, A. (2015). Extracellular matrix and growth factor engineering for controlled angiogenesis in regenerative medicine. Frontiers in bioengineering and bio- technology, 3, 45.
256. Li, H., Fredriksson, L., Li, X., & Eriksson, U. (2003). PDGF-D is a potent transforming and angiogenic growth factor. Onco- gene, 22(10), 1501-1510.
257. Cao, R., Bråkenhielm, E., Pawliuk, R., Wariaro, D., Post, M. J., Wahlberg, E., ... & Cao, Y. (2003). Angiogenic synergism, vascular stability and improvement of hind-limb ischemia by a combination of PDGF-BB and FGF-2. Nature medicine, 9(5), 604-613.
258. Apte, R. S., Chen, D. S., & Ferrara, N. (2019). VEGF in sig- naling and disease: beyond discovery and development. Cell, 176(6), 1248-1264.
259. Martínez, C. E., Smith, P. C., & Palma Alvarado, V. A. (2015). The influence of platelet-derived products on angiogenesis and tissue repair: a concise update. Frontiers in physiology, 6, 290.
260. Kemp, S. S., Lin, P. K., Sun, Z., Castaño, M. A., Yrigoin, K., Penn, M. R., & Davis, G. E. (2022). Molecular basis for peri- cyte-induced capillary tube network assembly and maturation. Frontiers in Cell and Developmental Biology, 10, 943533.
261. Stratman, A. N., & Davis, G. E. (2012). Endothelial cell-peri- cyte interactions stimulate basement membrane matrix assem- bly: influence on vascular tube remodeling, maturation, and stabilization. Microscopy and Microanalysis, 18(1), 68-80.
262. Holmes, D., Fitzgerald, P., Goldberg, S., LaBlanche, J. M., Lincoff, A. M., Savage, M., ... & Poland, M. (2000). The PRESTO (Prevention of restenosis with tranilast and its out- comes) protocol: a double-blind, placebo-controlled trial. American heart journal, 139(1), 23-31.
263. Stratman, A. N., Schwindt, A. E., Malotte, K. M., & Davis,
G. E. (2010). Endothelial-derived PDGF-BB and HB-EGF coordinately regulate pericyte recruitment during vasculogen- ic tube assembly and stabilization. Blood, The Journal of the American Society of Hematology, 116(22), 4720-4730.
264. Goumans, M. J., & Ten Dijke, P. (2018). TGF-β signaling in control of cardiovascular function. Cold Spring Harbor per- spectives in biology, 10(2), a022210.
265. Moriya, J., Wu, X., Zavala-Solorio, J., Ross, J., Liang, X. H., & Ferrara, N. (2014). Platelet-derived growth factor C pro- motes revascularization in ischemic limbs of diabetic mice. Journal of vascular surgery, 59(5), 1402-1409.
266. Shah, P., Keppler, L., & Rutkowski, J. (2014). A review of platelet derived growth factor playing pivotal role in bone re- generation. Journal of Oral Implantology, 40(3), 330-340.
267. Raza, S. L., & Cornelius, L. A. (2000, December). Matrix met- alloproteinases: pro-and anti-angiogenic activities. In Journal of Investigative Dermatology Symposium Proceedings (Vol. 5, No. 1, pp. 47-54). Elsevier.
268. Rundhaug, J. E. (2005). Matrix metalloproteinases and angi- ogenesis. Journal of cellular and molecular medicine, 9(2), 267-285.
269. Chantrain, C. F., Henriet, P., Jodele, S., Emonard, H., Feron, O., Courtoy, P. J., ... & Marbaix, E. (2006). Mechanisms of pericyte recruitment in tumour angiogenesis: a new role for metalloproteinases. European Journal of Cancer, 42(3), 310- 318.
270. Saunders, W. B., Bohnsack, B. L., Faske, J. B., Anthis, N. J., Bayless, K. J., Hirschi, K. K., & Davis, G. E. (2006). Coregu- lation of vascular tube stabilization by endothelial cell TIMP- 2 and pericyte TIMP-3. The Journal of cell biology, 175(1), 179-191.
271. Dean, E. W., Udelsman, B., & Breuer, C. K. (2012). Current advances in the translation of vascular tissue engineering to the treatment of pediatric congenital heart disease. The Yale journal of biology and medicine, 85(2), 229.
272. Wanjare, M., Kusuma, S., & Gerecht, S. (2014). Defining dif- ferences among perivascular cells derived from human pluri- potent stem cells. Stem Cell Reports, 2(5), 561-575.
273. Gerhardt, H., & Betsholtz, C. (2003). Endothelial-pericyte interactions in angiogenesis. Cell and tissue research, 314, 15-23.
274. Huang, H., Bhat, A., Woodnutt, G., & Lappe, R. (2010). Tar- geting the ANGPT–TIE2 pathway in malignancy. Nature Re- views Cancer, 10(8), 575-585.
275. Costa, S., Ragusa, M. A., Lo Buglio, G., Scilabra, S. D., & Nicosia, A. (2022). The repertoire of tissue inhibitors of met- alloproteases: evolution, regulation of extracellular matrix proteolysis, engineering and therapeutic challenges. Life, 12(8), 1145.
276. Wang, X., & Khalil, R. A. (2018). Matrix metalloproteinases, vascular remodeling, and vascular disease. Advances in phar- macology, 81, 241-330.
277. Marchand, M., Monnot, C., Muller, L., & Germain, S. (2019, May). Extracellular matrix scaffolding in angiogenesis and capillary homeostasis. In Seminars in cell & developmental biology (Vol. 89, pp. 147-156). Academic Press.
278. Davis, G. E., & Kemp, S. S. (2023). Extracellular matrix reg- ulation of vascular morphogenesis, maturation, and stabili- zation. Cold Spring Harbor perspectives in medicine, 13(4), a041156.
279. Kasravi, M., Ahmadi, A., Babajani, A., Mazloomnejad, R., Hatamnejad, M. R., Shariatzadeh, S., ... & Niknejad, H. (2023). Immunogenicity of decellularized extracellular matrix scaffolds: a bottleneck in tissue engineering and regenerative medicine. Biomaterials research, 27(1), 10.
280. Davis, G. E., & Senger, D. R. (2005). Endothelial extracel- lular matrix: biosynthesis, remodeling, and functions during vascular morphogenesis and neovessel stabilization. Circula- tion research, 97(11), 1093-1107.
281. Thomsen, M. S., Routhe, L. J., & Moos, T. (2017). The vascu- lar basement membrane in the healthy and pathological brain. Journal of Cerebral Blood Flow & Metabolism, 37(10), 3300-3317.
282. Pozzi, A., Yurchenco, P. D., & Iozzo, R. V. (2017). The na- ture and biology of basement membranes. Matrix Biology, 57, 1-11.
283. Mecham, R. P., & Ramirez, F. (2018). Extracellular determi- nants of arterial morphogenesis, growth, and homeostasis. Current topics in developmental biology, 130, 193-216.
284. Hill, M. A., & Meininger, G. A. (2012). Arteriolar vascular smooth muscle cells: mechanotransducers in a complex en- vironment. The international journal of biochemistry & cell biology, 44(9), 1505-1510.
285. Majesky, M. W., & Weiser-Evans, M. C. (2022). The adven- titia in arterial development, remodeling, and hypertension. Biochemical pharmacology, 205, 115259.
286. Tinajero, M. G., & Gotlieb, A. I. (2020). Recent developments in vascular adventitial pathobiology: the dynamic adventitia as a complex regulator of vascular disease. The American journal of pathology, 190(3), 520-534.
287. Stenmark, K. R., Yeager, M. E., El Kasmi, K. C., Nozik-Gray- ck, E., Gerasimovskaya, E. V., Li, M., ... & Frid, M. G. (2013). The adventitia: essential regulator of vascular wall structure and function. Annual review of physiology, 75(1), 23-47.
288. Majesky, M. W., Dong, X. R., Hoglund, V., Mahoney Jr, W. M., & Daum, G. (2011). The adventitia: a dynamic interface containing resident progenitor cells. Arteriosclerosis, throm- bosis, and vascular biology, 31(7), 1530-1539.
289. Eriksson, E. E. (2011). Intravital microscopy on atherosclero- sis in apolipoprotein E–deficient mice establishes microves- sels as major entry pathways for leukocytes to advanced le- sions. Circulation, 124(19), 2129-2138.
290. Maiellaro, K., & Taylor, W. R. (2007). The role of the adventi- tia in vascular inflammation. Cardiovascular research, 75(4), 640-648.
291. Rademakers, T., Douma, K., Hackeng, T. M., Post, M. J., Sluimer, J. C., Daemen, M. J., ... & van Zandvoort, M. A. (2013). Plaque-associated vasa vasorum in aged apolipopro- tein e–deficient mice exhibit proatherogenic functional fea- tures in vivo. Arteriosclerosis, thrombosis, and vascular biol- ogy, 33(2), 249-256.
292. Mazurek, R., Dave, J. M., Chandran, R. R., Misra, A., Sheikh, A. Q., & Greif, D. M. (2017). Vascular cells in blood vessel wall development and disease. Advances in Pharmacology, 78, 323-350.
293. Jain, M., & Chauhan, A. K. (2022). Role of integrins in modu- lating smooth muscle cell plasticity and vascular remodeling: from expression to therapeutic implications. Cells, 11(4), 646.
294. Déglise, S., Bechelli, C., & Allagnat, F. (2023). Vascular smooth muscle cells in intimal hyperplasia, an update. Fron- tiers in physiology, 13, 1081881.
295. Rifkin, D., Sachan, N., Singh, K., Sauber, E., Tellides, G., & Ramirez, F. (2022). The role of LTBPs in TGF beta signaling. Developmental Dynamics, 251(1), 75-84.
296. Crisan, M., Corselli, M., Chen, W. C., & Péault, B. (2012). Perivascular cells for regenerative medicine. Journal of cellu- lar and molecular medicine, 16(12), 2851-2860.
297. Sluimer, J. C., Kolodgie, F. D., Bijnens, A. P., Maxfield, K.,Pacheco, E., Kutys, B., ... & Daemen, M. J. (2009). Thin- walled microvessels in human coronary atherosclerotic plaques show incomplete endothelial junctions: relevance of compromised structural integrity for intraplaque microvascu- lar leakage. Journal of the American College of Cardiology, 53(17), 1517-1527.
298. Wang, M., Jiang, L., Monticone, R. E., & Lakatta, E. G. (2014). Proinflammation: the key to arterial aging. Trends in Endocrinology & Metabolism, 25(2), 72-79.
299. Wang, M., Monticone, R. E., & McGraw, K. R. (2020). Proin- flammation, profibrosis, and arterial aging. Aging Medicine, 3(3), 159-168.
300. Cui, N., Hu, M., & Khalil, R. A. (2017). Biochemical and bi- ological attributes of matrix metalloproteinases. Progress in molecular biology and translational science, 147, 1-73.
301. Rienks, M., Barallobre-Barreiro, J., & Mayr, M. (2018). The emerging role of the ADAMTS family in vascular diseases. Circulation research, 123(12), 1279-1281.
302. Fadini, G. P., Losordo, D., & Dimmeler, S. (2012). Critical reevaluation of endothelial progenitor cell phenotypes for therapeutic and diagnostic use. Circulation research, 110(4), 624-637.
303. Chen, F. M., Zhang, M., & Wu, Z. F. (2010). Toward delivery of multiple growth factors in tissue engineering. Biomaterials, 31(24), 6279-6308.
304. Rouwkema, J., Rivron, N. C., & van Blitterswijk, C. A. (2008). Vascularization in tissue engineering. Trends in bio- technology, 26(8), 434-441.
305. Zou, D., Zhang, Z., He, J., Zhang, K., Ye, D., Han, W., ... &
Jiang, X. (2012). Blood vessel formation in the tissue-engi- neered bone with the constitutively active form of HIF-1α me- diated BMSCs. Biomaterials, 33(7), 2097-2108.
306. Sun, G., Shen, Y. I., Kusuma, S., Fox-Talbot, K., Steenbergen, C. J., & Gerecht, S. (2011). Functional neovascularization of biodegradable dextran hydrogels with multiple angiogenic growth factors. Biomaterials, 32(1), 95-106.
307. Bai, Y., Bai, L., Zhou, J., Chen, H., & Zhang, L. (2018). Se- quential delivery of VEGF, FGF-2 and PDGF from the pol- ymeric system enhance HUVECs angiogenesis in vitro and CAM angiogenesis. Cellular Immunology, 323, 19-32.
308. Zamiri, P., Masli, S., Streilein, J. W., & Taylor, A. W. (2006). Pigment epithelial growth factor suppresses inflammation by modulating macrophage activation. Investigative ophthalmol- ogy & visual science, 47(9), 3912-3918.
309. Hao, X., Silva, E. A., Månsson-Broberg, A., Grinnemo, K. H., Siddiqui, A. J., Dellgren, G., ... & Sylvén, C. (2007). Angio- genic effects of sequential release of VEGF-A165 and PDGF- BB with alginate hydrogels after myocardial infarction. Car- diovascular research, 75(1), 178-185.
310. Chen, R. R., Silva, E. A., Yuen, W. W., & Mooney, D. J. (2007). Spatio–temporal VEGF and PDGF delivery patterns blood vessel formation and maturation. Pharmaceutical re- search, 24, 258-264.
311. Nillesen, S. T., Geutjes, P. J., Wismans, R., Schalkwijk, J., Daamen, W. F., & van Kuppevelt, T. H. (2007). Increased an-giogenesis and blood vessel maturation in acellular collagen– heparin scaffolds containing both FGF2 and VEGF. Biomate- rials, 28(6), 1123-1131.
312. Zieris, A., Prokoph, S., Levental, K. R., Welzel, P. B., Grim- mer, M., Freudenberg, U., & Werner, C. (2010). FGF-2 and VEGF functionalization of starPEG–heparin hydrogels to modulate biomolecular and physical cues of angiogenesis. Bi- omaterials, 31(31), 7985-7994.
313. Hori, Y., Ito, K., Hamamichi, S., Ozawa, Y., Matsui, J., Um- eda, I. O., & Fujii, H. (2017). Functional characterization of VEGF-and FGF-induced tumor blood vessel models in human cancer xenografts. Anticancer Research, 37(12), 6629-6638.
314. Khan, S., Villalobos, M. A., Choron, R. L., Chang, S., Brown,S. A., Carpenter, J. P., ... & Zhang, P. (2017). Fibroblast growth factor and vascular endothelial growth factor play a critical role in endotheliogenesis from human adipose-derived stem cells. Journal of Vascular Surgery, 65(5), 1483-1492.
315. Gavard, J., Patel, V., & Gutkind, J. S. (2008). Angiopoietin-1 prevents VEGF-induced endothelial permeability by seques- tering Src through mDia. Developmental cell, 14(1), 25-36.
316. Ngok, S. P., Geyer, R., Liu, M., Kourtidis, A., Agrawal, S., Wu, C., ... & Anastasiadis, P. Z. (2012). VEGF and Angiopoi- etin-1 exert opposing effects on cell junctions by regulating the Rho GEF Syx. Journal of Cell Biology, 199(7), 1103-1115.
317. Anisimov, A., Tvorogov, D., Alitalo, A., Leppänen, V. M., An, Y., Han, E. C., ... & Alitalo, K. (2013). Vascular endothelial growth factor-angiopoietin chimera with improved properties for therapeutic angiogenesis. Circulation, 127(4), 424-434.
318. Nissen, L. J., Cao, R., Hedlund, E. M., Wang, Z., Zhao, X., Wetterskog, D., ... & Cao, Y. (2007). Angiogenic factors FGF2 and PDGF-BB synergistically promote murine tumor neovas- cularization and metastasis. The Journal of clinical investiga- tion, 117(10), 2766-2777.
319. Gianni-Barrera, R., Di Maggio, N., Melly, L., Burger, M. G., Mujagic, E., Gürke, L., ... & Banfi, A. (2020). Therapeutic vascularization in regenerative medicine. Stem cells transla- tional medicine, 9(4), 433-444.
320. Gianni-Barrera, R., Burger, M., Wolff, T., Heberer, M., Schaefer, D. J., Gürke, L., ... & Banfi, A. (2016). Long-term safety and stability of angiogenesis induced by balanced sin- gle-vector co-expression of PDGF-BB and VEGF164 in skel- etal muscle. Scientific reports, 6(1), 21546.
321. Richardson, T. P., Peters, M. C., Ennett, A. B., & Mooney, D.J. (2001). Polymeric system for dual growth factor delivery.
Nature biotechnology, 19(11), 1029-1034.
322. Banfi, A., von Degenfeld, G., Gianni-Barrera, R., Reginato, S., Merchant, M. J., McDonald, D. M., & Blau, H. M. (2012). Therapeutic angiogenesis due to balanced single-vector deliv- ery of VEGF and PDGF-BB. The FASEB journal, 26(6), 2486.
323. Kang, D. H., Hughes, J., Mazzali, M., Schreiner, G. F., & Johnson, R. J. (2001). Impaired angiogenesis in the remnant kidney model: II. Vascular endothelial growth factor admin- istration reduces renal fibrosis and stabilizes renal function. Journal of the American Society of Nephrology, 12(7), 1448- 1457.
324. Kupatt, C., Hinkel, R., Pfosser, A., El-Aouni, C., Wuchrer, A., Fritz, A., ... & Boekstegers, P. (2010). Cotransfection of vascular endothelial growth factor-A and platelet-derived growth factor-B via recombinant adeno-associated virus re- solves chronic ischemic malperfusion: role of vessel matura- tion. Journal of the American College of Cardiology, 56(5), 414-422.
325. Salmerón-Sánchez, M., & Dalby, M. J. (2016). Synergistic growth factor microenvironments. Chemical Communica- tions, 52(91), 13327-13336.
326. Cao, L., Arany, P. R., Wang, Y. S., & Mooney, D. J. (2009).
Promoting angiogenesis via manipulation of VEGF respon- siveness with notch signaling. Biomaterials, 30(25), 4085- 4093.
327. Ramirez, F., & Rifkin, D. B. (2003). Cell signaling events: a view from the matrix. Matrix biology, 22(2), 101-107.
328. Martino, M. M., Tortelli, F., Mochizuki, M., Traub, S., Ben-David, D., Kuhn, G. A., ... & Hubbell, J. A. (2011). Engi- neering the growth factor microenvironment with fibronectin domains to promote wound and bone tissue healing. Science translational medicine, 3(100), 100ra89-100ra89.
329. Bray, L. J., Binner, M., Holzheu, A., Friedrichs, J., Freuden- berg, U., Hutmacher, D. W., & Werner, C. (2015). Multi-para- metric hydrogels support 3D in vitro bioengineered microen- vironment models of tumour angiogenesis. Biomaterials, 53, 609-620.
330. García, J. R., Clark, A. Y., & García, A. J. (2016). Integrinâ? specific hydrogels functionalized with VEGF for vasculariza- tion and bone regeneration of criticalâ?size bone defects. Jour- nal of biomedical materials research Part A, 104(4), 889-900.
331. Llopis-Hernández, V., Cantini, M., González-García, C., Cheng, Z. A., Yang, J., Tsimbouri, P. M., ... & Salm- erón-Sánchez, M. (2016). Material-driven fibronectin assem- bly for high-efficiency presentation of growth factors. Science advances, 2(8), e1600188.
332. Moulisová, V., Gonzalez-García, C., Cantini, M., Rod- rigo-Navarro, A., Weaver, J., Costell, M., ... & Salm- erón-Sánchez, M. (2017). Engineered microenvironments for synergistic VEGF–Integrin signalling during vascularization. Biomaterials, 126, 61-74.
333. Shekaran, A., García, J. R., Clark, A. Y., Kavanaugh, T. E., Lin, A. S., Guldberg, R. E., & García, A. J. (2014). Bone re- generation using an alpha 2 beta 1 integrin-specific hydrogel as a BMP-2 delivery vehicle. Biomaterials, 35(21), 5453- 5461.
334. Schneller, M., Vuori, K., & Ruoslahti, E. (1997). αvβ3 inte- grin associates with activated insulin and PDGFβ receptors and potentiates the biological activity of PDGF. The EMBO journal.
335. Scaffidi, A. K., Petrovic, N., Moodley, Y. P., Fogel-Petrovic, M., Kroeger, K. M., Seeber, R. M., ... & Knight, D. A. (2004). αvβ3 Integrin interacts with the transforming growth factor β (TGFβ) type II receptor to potentiate the proliferative effects of TGFβ1 in living human lung fibroblasts. Journal of Biolog- ical Chemistry, 279(36), 37726-37733.
336. Mattila, E., Pellinen, T., Nevo, J., Vuoriluoto, K., Arjonen, A., & Ivaska, J. (2005). Negative regulation of EGFR signalling through integrin-a 1 ß 1-mediated activation of protein tyros- ine phosphatase TCPTP. Nature Cell Biology, 7(1).
337. Mattila, E., Marttila, H., Sahlberg, N., Kohonen, P., TaÌ?htinen, S., Halonen, P., ... & Ivaska, J. (2010). Inhibition of receptor tyrosine kinase signalling by small molecule agonist of T-cell protein tyrosine phosphatase. BMC cancer, 10, 1-12.
338. Monteiro A, I., Kollmetz, T., & Malmström, J. (2018). En- gineered systems to study the synergistic signaling between integrin-mediated mechanotransduction and growth factors. Biointerphases, 13(6).
339. Lamalice, L., Le Boeuf, F., & Huot, J. (2007). Endothelial cell migration during angiogenesis. Circulation research, 100(6), 782-794.
340. Tabas, I., Williams, K. J., & Borén, J. (2007). Subendothelial lipoprotein retention as the initiating process in atherosclero- sis: update and therapeutic implications. Circulation, 116(16), 1832-1844.
341. Nielsen, L. B., Grønholdt, M. L., Schroeder, T. V., Stender, S., & Nordestgaard, B. G. (1997). In vivo transfer of lipoprotein
(a) into human atherosclerotic carotid arterial intima. Arteri- osclerosis, thrombosis, and vascular biology, 17(5), 905-911.
342. Bartels, E. D., Christoffersen, C., Lindholm, M. W., & Niels- en, L. B. (2015). Altered metabolism of LDL in the arteri- al wall precedes atherosclerosis regression. Circulation re- search, 117(11), 933-942.
343. Ference, B. A., Ginsberg, H. N., Graham, I., Ray, K. K., Pack-ard, C. J., Bruckert, E., ... & Catapano, A. L. (2017). Low-den- sity lipoproteins cause atherosclerotic cardiovascular disease.
1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis So- ciety Consensus Panel. European heart journal, 38(32), 2459- 2472.
344. Nordestgaard, B. G., Chapman, M. J., Humphries, S. E., Ginsberg, H. N., Masana, L., Descamps, O. S., ... & Europe- an Atherosclerosis Society Consensus Panel. (2013). Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the Europe- an Atherosclerosis Society. European heart journal, 34(45), 3478-3490.
345. Nordestgaard, B. G., Wootton, R., & Lewis, B. (1995). Selec- tive retention of VLDL, IDL, and LDL in the arterial intima of genetically hyperlipidemic rabbits in vivo: molecular size as a determinant of fractional loss from the intima–inner me- dia. Arteriosclerosis, thrombosis, and vascular biology, 15(4), 534-542.
346. Simons, M. (2021). Fibroblast growth factors: The keepers of endothelial normalcy. The Journal of Clinical Investigation, 131(17).
347. Chen, P. Y., Qin, L., Baeyens, N., Li, G., Afolabi, T., Budatha, M., ... & Simons, M. (2015). Endothelial-to-mesenchymal transition drives atherosclerosis progression. The Journal of clinical investigation, 125(12), 4514-4528.
348. Soehnlein, O. (2012). Multiple roles for neutrophils in athero- sclerosis. Circulation research, 110(6), 875-888.
349. Soehnlein, O., Lindbom, L., & Weber, C. (2009). Mecha- nisms underlying neutrophil-mediated monocyte recruitment. Blood, The Journal of the American Society of Hematology, 114(21), 4613-4623.
350. Chèvre, R., González-Granado, J. M., Megens, R. T., Sreer- amkumar, V., Silvestre-Roig, C., Molina-Sánchez, P., ... & Andrés, V. (2014). High-resolution imaging of intravascular atherogenic inflammation in live mice. Circulation research, 114(5), 770-779.
351. Sundd, P., Gutierrez, E., Koltsova, E. K., Kuwano, Y., Fukuda, S., Pospieszalska, M. K., ... & Ley, K. (2012). ‘Slings’ enable neutrophil rolling at high shear. Nature, 488(7411), 399-403.
352. Muller, W. A. (2013). Getting leukocytes to the site of inflam- mation. Veterinary pathology, 50(1), 7-22.
353. Sumagin, R., Prizant, H., Lomakina, E., Waugh, R. E., & Sarelius, I. H. (2010). LFA-1 and Mac-1 define characteristi- cally different intralumenal crawling and emigration patterns for monocytes and neutrophils in situ. The Journal of Immu- nology, 185(11), 7057-7066.
354. Fan, Z., McArdle, S., Marki, A., Mikulski, Z., Gutierrez, E., Engelhardt, B., ... & Ley, K. (2016). Neutrophil recruitment limited by high-affinity bent β2 integrin binding ligand in cis. Nature communications, 7(1), 12658.
355. Barreiro, O., Yáñez-Mó, M., Serrador, J. M., Montoya, M. C., Vicente-Manzanares, M., Tejedor, R., ... & Sánchez-Madrid,
F. (2002). Dynamic interaction of VCAM-1 and ICAM-1 with moesin and ezrin in a novel endothelial docking structure for adherent leukocytes. Journal of Cell Biology, 157(7), 1233- 1245.
356. Carman, C. V., & Springer, T. A. (2004). A transmigratory cup in leukocyte diapedesis both through individual vascular endothelial cells and between them. Journal of Cell Biology, 167(2), 377-388.
357. Barzilai, S., Yadav, S. K., Morrell, S., Roncato, F., Klein, E., Stoler-Barak, L., ... & Alon, R. (2017). Leukocytes breach en- dothelial barriers by insertion of nuclear lobes and disassem- bly of endothelial actin filaments. Cell reports, 18(3), 685- 699.
358. Sluiter, T. J., van Buul, J. D., Huveneers, S., Quax, P. H., & de Vries, M. R. (2021). Endothelial barrier function and leu- kocyte transmigration in atherosclerosis. Biomedicines, 9(4), 328.
359. Nourshargh, S., Hordijk, P. L., & Sixt, M. (2010). Breaching multiple barriers: leukocyte motility through venular walls and the interstitium. Nature reviews Molecular cell biology, 11(5), 366-378.
360. Maas, S. L., Soehnlein, O., & Viola, J. R. (2018). Organ-spe- cific mechanisms of transendothelial neutrophil migration in the lung, liver, kidney, and aorta. Frontiers in immunology, 9, 2739.
361. Salminen, A. T., Allahyari, Z., Gholizadeh, S., McCloskey, M. C., Ajalik, R., Cottle, R. N., ... & McGrath, J. L. (2020). In vitro studies of transendothelial migration for biological and drug discovery. Frontiers in Medical Technology, 2, 600616.
362. Pezhman, L., Tahrani, A., & Chimen, M. (2021). Dysregula-tion of leukocyte trafficking in type 2 diabetes: mechanisms and potential therapeutic avenues. Frontiers in cell and devel- opmental biology, 9, 624184.
363. Groyer, E., Caligiuri, G., Laschet-Khallou, J., & Nicoletti,A. (2006). Immunological aspects of atherosclerosis. Presse Medicale (Paris, France: 1983), 35(3 Pt 2), 475-486.
364. Zernecke, A., Winkels, H., Cochain, C., Williams, J. W., Wolf, D., Soehnlein, O., ... & Ley, K. (2020). Meta-analysis of leu- kocyte diversity in atherosclerotic mouse aortas. Circulation research, 127(3), 402-426.
365. Jessup, W., & Kritharides, L. (2000). Metabolism of oxidized LDL by macrophages. Current opinion in lipidology, 11(5), 473-481.
366. Sorci-Thomas, M. G., & Thomas, M. J. (2016). Microdo- mains, inflammation, and atherosclerosis. Circulation re- search, 118(4), 679-691.
367. Zuchtriegel, G., Uhl, B., Hessenauer, M. E., Kurz, A. R., Re- hberg, M., Lauber, K., ... & Reichel, C. A. (2015). Spatiotem- poral expression dynamics of selectins govern the sequential extravasation of neutrophils and monocytes in the acute in- flammatory response. Arteriosclerosis, thrombosis, and vas- cular biology, 35(4), 899-910.
368. Nourshargh, S., & Alon, R. (2014). Leukocyte migration into inflamed tissues. Immunity, 41(5), 694-707.
369. Poznyak, A. V., Nikiforov, N. G., Markin, A. M., Kashirskikh,D. A., Myasoedova, V. A., Gerasimova, E. V., & Orekhov, A.N. (2021). Overview of OxLDL and its impact on cardiovas- cular health: focus on atherosclerosis. Frontiers in Pharma- cology, 11, 613780.
370. Endemann, G., Stanton, L. W., Madden, K. S., Bryant, C. M., White, R. T., & Protter, A. A. (1993). CD36 is a receptor for oxidized low density lipoprotein. Journal of Biological Chem- istry, 268(16), 11811-11816.
371. Acton, S. L., Scherer, P. E., Lodish, H. F., & Krieger, M. (1994). Expression cloning of SR-BI, a CD36-related class B scavenger receptor. Journal of Biological Chemistry, 269(33), 21003-21009.
372. Duewell, P., Kono, H., Rayner, K. J., Sirois, C. M., Vladimer, G., Bauernfeind, F. G., ... & Latz, E. (2010). NLRP3 inflam- masomes are required for atherogenesis and activated by cho- lesterol crystals. Nature, 464(7293), 1357-1361.
373. Navab, M., Ananthramaiah, G. M., Reddy, S. T., Van Lenten,B. J., Ansell, B. J., Fonarow, G. C., ... & Fogelman, A. M. (2004). Thematic review series: the pathogenesis of athero- sclerosis the oxidation hypothesis of atherogenesis: the role of oxidized phospholipids and HDL. Journal of lipid research, 45(6), 993-1007.
374. Hashimoto, K., Kataoka, N., Nakamura, E., Tsujioka, K., & Kajiya, F. (2007). Oxidized LDL specifically promotes the in- itiation of monocyte invasion during transendothelial migra- tion with upregulated PECAM-1 and downregulated VE-cad- herin on endothelial junctions. Atherosclerosis, 194(2), e9-e17.
375. Brugger, W., Kreutz, M., & Andreesen, R. (1991). Mac- rophage colony stimulating factor is required for human monocyte survival and acts as a cofactor for their terminal differentiation to macrophages in vitro. Journal of leukocyte biology, 49(5), 483-488.
376. Lehtonen, A., Matikainen, S., Miettinen, M., & Julkunen, I. (2002). Granulocyte-macrophage colony-stimulating factor (GM-CSF)-induced STAT5 activation and target-gene ex- pression during human monocyte/macrophage differentiation. Journal of leukocyte biology, 71(3), 511-519.
377. Davis, G. E., Thomas, J. S., & Madden, S. (1997). The α4β1 integrin can mediate leukocyte adhesion to casein and dena- tured protein substrates. Journal of leukocyte biology, 62(3), 318-328.
378. A. Tedgui, Z. Mallat, (1999). Atherosclerotic plaque forma- tion, Rev. Prat. 49.
379. Zhang, X., Sessa, W. C., & Fernández-Hernando, C. (2018). Endothelial transcytosis of lipoproteins in atherosclerosis. Frontiers in cardiovascular medicine, 5, 130.
380. Kim, K. W., Ivanov, S., & Williams, J. W. (2020). Monocyte recruitment, specification, and function in atherosclerosis. Cells, 10(1), 15.
381. BaÌ?ck, M., Yurdagul Jr, A., Tabas, I., Öörni, K., & Kovanen, P.T. (2019). Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities. Nature Reviews Car- diology, 16(7), 389-406.
382. Que, X., Hung, M. Y., Yeang, C., Gonen, A., Prohaska, T. A., Sun, X., ... & Witztum, J. L. (2018). Oxidized phospholipids are proinflammatory and proatherogenic in hypercholestero- laemic mice. Nature, 558(7709), 301-306.
383. Mourouzis, K., Siasos, G., Oikonomou, E., Zaromitidou, M., Tsigkou, V., Antonopoulos, A., ... & Tousoulis, D. (2021). Lipoprotein-associated phospholipase A2 levels, endothelial dysfunction and arterial stiffness in patients with stable coro- nary artery disease. Lipids in health and disease, 20, 1-9.
384. Bennett, M. R., Sinha, S., & Owens, G. K. (2016). Vascular smooth muscle cells in atherosclerosis. Circulation research, 118(4), 692-702.
385. Llorente-Cortés, V., MartıÌnez-González, J., & Badimon, L. (2000). LDL receptor–related protein mediates uptake of ag- gregated LDL in human vascular smooth muscle cells. Arte- riosclerosis, thrombosis, and vascular biology, 20(6), 1572- 1579.
386. Saunders, W. B., Bayless, K. J., & Davis, G. E. (2005). MMP- 1 activation by serine proteases and MMP-10 induces human capillary tubular network collapse and regression in 3D col- lagen matrices. Journal of cell science, 118(10), 2325-2340.
387. Bolshakov, I. N., Gornostaev, L. M., Fominykh, O. I., & Svet- lakov, A. V. (2022). Synthesis, chemical and biomedical as- pects of the use of sulfated chitosan. Polymers, 14(16), 3431.
388. Tabas, I., & Bornfeldt, K. E. (2016). Macrophage phenotype and function in different stages of atherosclerosis. Circulation research, 118(4), 653-667.
389. Bentzon, J. F., Otsuka, F., Virmani, R., & Falk, E. (2014). Mechanisms of plaque formation and rupture. Circulation re- search, 114(12), 1852-1866.
390. Visse, R., & Nagase, H. (2003). Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, func-tion, and biochemistry. Circulation research, 92(8), 827-839.
391. Kumar, B. V., Connors, T. J., & Farber, D. L. (2018). Human T cell development, localization, and function throughout life. Immunity, 48(2), 202-213.
392. Zhou, X., Nicoletti, A., Elhage, R., & Hansson, G. K. (2000). Transfer of CD4+ T cells aggravates atherosclerosis in im- munodeficient apolipoprotein E knockout mice. Circulation, 102(24), 2919-2922.
393. Zhou, X., Robertson, A. K. L., Hjerpe, C., & Hansson, G. K. (2006). Adoptive transfer of CD4+ T cells reactive to modified low-density lipoprotein aggravates atherosclerosis. Arterio- sclerosis, thrombosis, and vascular biology, 26(4), 864-870.
394. Zhou, X., Robertson, A. K. L., Rudling, M., Parini, P., & Hansson, G. K. (2005). Lesion development and response to immunization reveal a complex role for CD4 in atherosclero- sis. Circulation research, 96(4), 427-434.
395. Li, J., & Ley, K. (2015). Lymphocyte migration into ather- osclerotic plaque. Arteriosclerosis, thrombosis, and vascular biology, 35(1), 40-49.
396. Foks, A. C., Lichtman, A. H., & Kuiper, J. (2015). Treat- ing atherosclerosis with regulatory T cells. Arteriosclerosis, thrombosis, and vascular biology, 35(2), 280-287.
397. Proto, J. D., Doran, A. C., Gusarova, G., Yurdagul, A., Sozen, E., Subramanian, M., ... & Tabas, I. (2018). Regulatory T cells promote macrophage efferocytosis during inflammation reso- lution. Immunity, 49(4), 666-677.
398. Ley, K. (2020). Role of the adaptive immune system in ath- erosclerosis. Biochemical Society Transactions, 48(5), 2273-
2281.
399. MacRitchie, N., Grassia, G., Noonan, J., Cole, J. E., Hughes,
C. E., Schroeder, J., ... & Maffia, P. (2020). The aorta can act as a site of naive CD4+ T-cell priming. Cardiovascular re- search, 116(2), 306-316.
400. Matsumoto, M., Shigeta, A., Miyasaka, M., & Hirata, T. (2008). CD43 plays both antiadhesive and proadhesive roles in neutrophil rolling in a context-dependent manner. The Journal of Immunology, 181(5), 3628-3635.
401. Nácher, M., Blázquez, A. B., Shao, B., Matesanz, A., Proph- ete, C., Berin, M. C., ... & Hidalgo, A. (2011). Physiological contribution of CD44 as a ligand for E-selectin during inflam- matory T-cell recruitment. The American journal of patholo- gy, 178(5), 2437-2446.
402. Shulman, Z., Cohen, S. J., Roediger, B., Kalchenko, V., Jain, R., Grabovsky, V., ... & Alon, R. (2012). Transendothelial mi- gration of lymphocytes mediated by intraendothelial vesicle stores rather than by extracellular chemokine depots. Nature immunology, 13(1), 67-76.
403. Sage, A. P., Tsiantoulas, D., Binder, C. J., & Mallat, Z. (2019). The role of B cells in atherosclerosis. Nature Reviews Cardi- ology, 16(3), 180-196.
404. Wang, L., & Tang, C. (2020). Targeting platelet in athero- sclerosis plaque formation: current knowledge and future perspectives. International journal of molecular sciences, 21(24), 9760.
405. Magwenzi, S., Woodward, C., Wraith, K. S., Aburima, A., Raslan, Z., Jones, H., ... & Naseem, K. M. (2015). Oxidized LDL activates blood platelets through CD36/NOX2–mediat- ed inhibition of the cGMP/protein kinase G signaling cascade. Blood, The Journal of the American Society of Hematology, 125(17), 2693-2703.
406. Bakogiannis, C., Sachse, M., Stamatelopoulos, K., & Stellos,K. (2019). Platelet-derived chemokines in inflammation and
atherosclerosis. Cytokine, 122, 154157.
407. Shevchuk, O., Begonja, A. J., Gambaryan, S., Totzeck, M., Rassaf, T., Huber, T. B., ... & Sickmann, A. (2021). Proteom- ics: a tool to study platelet function. International journal of molecular sciences, 22(9), 4776.
408. Barrett, T. J., Schlegel, M., Zhou, F., Gorenchtein, M., Bol- storff, J., Moore, K. J., ... & Berger, J. S. (2019). Platelet reg- ulation of myeloid suppressor of cytokine signaling 3 acceler- ates atherosclerosis. Science translational medicine, 11(517), eaax0481.
409. Mantovani, A., Sica, A., Sozzani, S., Allavena, P., Vecchi, A., & Locati, M. (2004). The chemokine system in diverse forms of macrophage activation and polarization. Trends in immu- nology, 25(12), 677-686.
410. Martinez, F. O., Sica, A., Mantovani, A., & Locati, M. (2008). Macrophage activation and polarization. Front biosci, 13(13), 453-461.
411. Meza, D., Shanmugavelayudam, S. K., Mendoza, A., Sanchez, C., Rubenstein, D. A., & Yin, W. (2017). Platelets modulate endothelial cell response to dynamic shear stress through PE- CAM-1. Thrombosis Research, 150, 44-50.
412. Tersteeg, C., Heijnen, H. F., Eckly, A., Pasterkamp, G., Ur- banus, R. T., Maas, C., ... & Roest, M. (2014). FLow-Induced PRotrusions (FLIPRs) a platelet-derived platform for the re- trieval of microparticles by monocytes and neutrophils. Cir- culation research, 114(5), 780-791.
413. Baidžajevas, K., Hadadi, É., Lee, B., Lum, J., Shihui, F., Sud- bery, I., ... & Wilson, H. L. (2020). Macrophage polarisation associated with atherosclerosis differentially affects their ca- pacity to handle lipids. Atherosclerosis, 305, 10-18.
414. Busch, M., Westhofen, T. C., Koch, M., Lutz, M. B., & Ze- rnecke, A. (2014). Dendritic cell subset distributions in the aorta in healthy and atherosclerotic mice. PLoS One, 9(2), e88452.
415. Choi, J. H., Do, Y., Cheong, C., Koh, H., Boscardin, S. B., Oh, Y. S., ... & Steinman, R. M. (2009). Identification of an- tigen-presenting dendritic cells in mouse aorta and cardiac valves. Journal of Experimental Medicine, 206(3), 497-505.
416. Sichien, D., Lambrecht, B. N., Guilliams, M., & Scott, C. L. (2017). Development of conventional dendritic cells: from common bone marrow progenitors to multiple subsets in pe- ripheral tissues. Mucosal immunology, 10(4), 831-844.
417. Liu, J., Zhang, X., Cheng, Y., & Cao, X. (2021). Dendritic cell migration in inflammation and immunity. Cellular & molecu- lar immunology, 18(11), 2461-2471.
418. Tiberio, L., Del Prete, A., Schioppa, T., Sozio, F., Bosisio, D., & Sozzani, S. (2018). Chemokine and chemotactic signals in dendritic cell migration. Cellular & molecular immunology, 15(4), 346-352.
419. Wang, L., Gao, B., Wu, M., Yuan, W., Liang, P., & Huang, J.(2021). Profiles of immune cell infiltration in carotid artery atherosclerosis based on gene expression data. Frontiers in Immunology, 12, 599512.
420. Han, H., Du, R., Cheng, P., Zhang, J., Chen, Y., & Li, G. (2021). Comprehensive analysis of the immune infiltrates and aberrant pathways activation in atherosclerotic plaque. Fron- tiers in Cardiovascular Medicine, 7, 602345.
421. Cortenbach, K. R., Cano, D. M., Meek, J., Gorris, M. A.,
Staal, A. H., Srinivas, M., ... & van Kimmenade, R. R. (2023). Topography of immune cell infiltration in different stages of coronary atherosclerosis revealed by multiplex immunohisto- chemistry. IJC Heart & Vasculature, 44, 101111.
422. Gotsman, I., Sharpe, A. H., & Lichtman, A. H. (2008). T-cell costimulation and coinhibition in atherosclerosis. Circulation research, 103(11), 1220-1231.
423. Britsch, S., Langer, H., Duerschmied, D., & Becher, T. (2024). The evolving role of dendritic cells in atherosclerosis. Inter- national Journal of Molecular Sciences, 25(4), 2450.
424. Young, J. L., Libby, P., & Schönbeck, U. (2002). Cytokines in the pathogenesis of atherosclerosis. Thrombosis and haemo- stasis, 88(10), 554-567.
425. Lee, K., Silva, E. A., & Mooney, D. J. (2011). Growth factor delivery-based tissue engineering: general approaches and a review of recent developments. Journal of the Royal Society Interface, 8(55), 153-170.
426. Hynes, R. O. (2009). The extracellular matrix: not just pretty fibrils. Science, 326(5957), 1216-1219.
427. Paralkar, V. M., Vukicevic, S., & Reddi, A. H. (1991). Trans- forming growth factor β type 1 binds to collagen IV of base- ment membrane matrix: Implications for development. Devel- opmental biology, 143(2), 303-308.
428. Martino, M. M., Briquez, P. S., Ranga, A., Lutolf, M. P., & Hubbell, J. A. (2013). Heparin-binding domain of fibrin (ogen) binds growth factors and promotes tissue repair when incorporated within a synthetic matrix. Proceedings of the Na- tional Academy of Sciences, 110(12), 4563-4568.
429. Martino, M. M., & Hubbell, J. A. (2010). The 12th–14th type III repeats of fibronectin function as a highly promiscuous growth factorâ?binding domain. The FASEB Journal, 24(12), 4711-4721.
430. Wijelath, E. S., Rahman, S., Namekata, M., Murray, J., Nishi- mura, T., Mostafavi-Pour, Z., ... & Sobel, M. (2006). Hepa- rin-II domain of fibronectin is a vascular endothelial growth factor-binding domain: enhancement of VEGF biological ac- tivity by a singular growth factor/matrix protein synergism. Circulation research, 99(8), 853-860.
431. Kricker, J. A., Towne, C. L., Firth, S. M., Herington, A. C., & Upton, Z. (2003). Structural and functional evidence for the interaction of insulin-like growth factors (IGFs) and IGF binding proteins with vitronectin. Endocrinology, 144(7), 2807-2815.
432. Hallmann, R., Zhang, X., Di Russo, J., Li, L., Song, J., Han- nocks, M. J., & Sorokin, L. (2015). The regulation of immune cell trafficking by the extracellular matrix. Current opinion in cell biology, 36, 54-61.
433. Friedl, P., & Mayor, R. (2017). Tuning collective cell migration by cell–cell junction regulation. Cold Spring Harbor per- spectives in biology, 9(4), a029199.
434. Park, J. E., Keller, G. A., & Ferrara, N. (1993). The vascular endothelial growth factor (VEGF) isoforms: differential depo- sition into the subepithelial extracellular matrix and bioactivi- ty of extracellular matrix-bound VEGF. Molecular biology of the cell, 4(12), 1317-1326.
435. Atanasova, M., & Whitty, A. (2012). Understanding cytokine and growth factor receptor activation mechanisms. Critical reviews in biochemistry and molecular biology, 47(6), 502- 530.
436. Rozario, T., & DeSimone, D. W. (2010). The extracellular matrix in development and morphogenesis: a dynamic view. Developmental biology, 341(1), 126-140.
437. Korpisalo, P., & YlaÌ?-Herttuala, S. (2010). Stimulation of func- tional vessel growth by gene therapy. Integrative Biology, 2(2- 3), 102-112.
438. Mussbacher, M., Schossleitner, K., Kral-Pointner, J. B., Salz- mann, M., Schrammel, A., & Schmid, J. A. (2022). More than just a monolayer: the multifaceted role of endothelial cells in the pathophysiology of atherosclerosis. Current atherosclero- sis reports, 24(6), 483-492.
439. Gimbrone Jr, M. A., & García-Cardeña, G. (2016). Endothe- lial cell dysfunction and the pathobiology of atherosclerosis. Circulation research, 118(4), 620-636.
440. Potente, M., Gerhardt, H., & Carmeliet, P. (2011). Basic and therapeutic aspects of angiogenesis. Cell, 146(6), 873-887.
441. Benita, Y., Kikuchi, H., Smith, A. D., Zhang, M. Q., Chung,
D. C., & Xavier, R. J. (2009). An integrative genomics ap- proach identifies Hypoxia Inducible Factor-1 (HIF-1)-target genes that form the core response to hypoxia. Nucleic acids research, 37(14), 4587-4602.
442. Ortiz-Barahona, A., Villar, D., Pescador, N., Amigo, J., & del Peso, L. (2010). Genome-wide identification of hypoxia-in- ducible factor binding sites and target genes by a probabilis- tic model integrating transcription-profiling data and in silico binding site prediction. Nucleic acids research, 38(7), 2332- 2345.
443. Semenza, G. L. (2009). Regulation of vascularization by hy- poxia inducible factor 1. Annals of the New York Academy of Sciences, 1177(1), 2-8.
444. Jain, T., Nikolopoulou, E. A., Xu, Q., & Qu, A. (2018). Hy- poxia inducible factor as a therapeutic target for atherosclero- sis. Pharmacology & therapeutics, 183, 22-33.
445. Rajagopalan, S., Olin, J., Deitcher, S., Pieczek, A., Laird, J., Grossman, P. M., ... & Chronos, N. (2007). Use of a constitu- tively active hypoxia-inducible factor-1α transgene as a ther- apeutic strategy in no-option critical limb ischemia patients: phase I dose-escalation experience. Circulation, 115(10), 1234-1243.
446. Kuan, C. Y., Chen, H. R., Gao, N., Kuo, Y. M., Chen, C. W.,Yang, D., ... & Sun, Y. Y. (2021). Brain-targeted hypoxia-in- ducible factor stabilization reduces neonatal hypoxic-ischem- ic brain injury. Neurobiology of disease, 148, 105200.
447. Nakashima, M., Watanabe, M., Nakano, K., Uchimaru, K., & Horie, R. (2021). Differentiation of Hodgkin lymphoma cells by reactive oxygen species and regulation by heme oxygen- aseâ?1 through HIFâ?1α. Cancer Science, 112(6), 2542-2555.
448. Rajendran, P., Rengarajan, T., Thangavel, J., Nishigaki, Y., Sakthisekaran, D., Sethi, G., & Nishigaki, I. (2013). The vas- cular endothelium and human diseases. International journal of biological sciences, 9(10), 1057.
449. Kalinin, R. E., Gryaznov, S. V., Nikiforov, A. A., Kamaev, A. A., Shvalb, A. P., & Slepnev, A. A. (2015). Nitric oxide synthase and endothelin-1 gene polymorphism in lower limb chronic venous insufficiency. IP Pavlov Russian Medical Bio- logical Herald, 23(4), 97-102.
450. Dow, C. A., Templeton, D. L., Lincenberg, G. M., Greiner,J. J., Stauffer, B. L., & DeSouza, C. A. (2016). Elevations in C-reactive protein and endothelin-1 system activity in hu- mans. Life sciences, 159, 66-70.
451. Trinity, J. D., Barrett-O’Keefe, Z., Ives, S. J., Morgan, G., Rossman, M. J., Donato, A. J., ... & Wray, D. W. (2016). En- dogenous endothelin-1 and femoral artery shear rate: impact of age and implications for atherosclerosis. Journal of hyper- tension, 34(2), 266-273.
452. B. A., Shelest. (2016). Peripheral vessel wall changes in hy- pertensive patients with gout, Ter. Arkh. 88 (5), 43-46.
453. Pavlides, S., Gutierrez-Pajares, J. L., Katiyar, S., Jasmin, J. F., Mercier, I., Walters, R., ... & Frank, P. G. (2014). Caveolin-1 regulates the anti-atherogenic properties of macrophages. Cell and tissue research, 358, 821-831.
454. Zborovskaya, I. B., Galetskiy, S. A., & Komel’kov, A. V. (2016). Microdomain forming proteins in oncogenesis. Ad- vances in molecular oncology, 3(3), 16-29.
455. Engelberger, R. P., Limacher, A., Kucher, N., Baumann, F., Silbernagel, G., Benghozi, R., ... & Baumgartner, I. (2015). Biological variation of established and novel biomarkers for atherosclerosis: Results from a prospective, parallel-group co- hort study. Clinica Chimica Acta, 447, 16-22.
456. Rozenberg, I., Sluka, S. H., Mocharla, P., Hallenberg, A., Rotzius, P., Boren, J., ... & Tanner, F. C. (2011). Deletion of L-selectin increases atherosclerosis development in ApoE−/− mice. PLoS One, 6(7), e21675.
457. I.S. Skopec, N.N. Vezikova, I.M. Marusenko, O.Y. Baryshe- va, A.V. Malafeev, A.N. Malygin, 12 (2016). Сorrelation of inflammation biomarkers with the traditional risk factors in patients with acute coronary syndrome, Rational Pharmaco- therapy in Cardiology. 12(2), 166-170.
458. Galkina, E., & Ley, K. (2007). Vascular adhesion molecules in atherosclerosis. Arteriosclerosis, thrombosis, and vascular biology, 27(11), 2292-2301.
459. Kitagawa, K., Matsumoto, M., Sasaki, T., Hashimoto, H., Ku- wabara, K., Ohtsuki, T., & Hori, M. (2002). Involvement of ICAM-1 in the progression of atherosclerosis in APOE-knock- out mice. Atherosclerosis, 160(2), 305-310.
460. Signorelli, S. S., Anzaldi, M., Libra, M., Navolanic, P. M., Malaponte, G., Mangano, K., ... & Neri, S. (2016). Plasma levels of inflammatory biomarkers in peripheral arterial dis- ease: results of a cohort study. Angiology, 67(9), 870-874.
461. I.S. Belokopytova, O.V. Moskaletz, F.N. Paleev, O.V. Zotova, 4 (2013). The diagnostic value of adhesive molecules sICAM -1 and sVCAM-1 in ischemic heart disease, The Journal of Atherosclerosis and Dyslipidemias.
462. Editors. (2013). Circulation Research Thematic Synopsis:Atherosclerosis. Circulation Research, 112(10), e118-e147.
463. Al-Ghurabi, M. E., Muhi, A. A., & Al-Mudhafar, D. H. (2015). Vascular cell adhesion molecule-1 and endothelial leukocyte adhesion molecule-1 as markers of atherosclerosis of NIDDM. Am J Life Sci, 3(1), 22-26.
464. Bala, G., Blykers, A., Xavier, C., Descamps, B., Broisat, A., Ghezzi, C., ... & Hernot, S. (2016). Targeting of vascular cell adhesion molecule-1 by 18F-labelled nanobodies for PET/CT imaging of inflamed atherosclerotic plaques. European Heart Journal-Cardiovascular Imaging, 17(9), 1001-1008.
465. Ni, W., Egashira, K., Kitamoto, S., Kataoka, C., Koyanagi, M., Inoue, S., ... & Takeshita, A. (2001). New anti–mono- cyte chemoattractant protein-1 gene therapy attenuates ath- erosclerosis in apolipoprotein E–knockout mice. Circulation, 103(16), 2096-2101.
466. Tsai, M. K., Hsieh, C. C., Kuo, H. F., Lee, M. S., Huang, M.
Y., Kuo, C. H., & Hung, C. H. (2015). Effect of prostaglandin I 2 analogs on monocyte chemoattractant protein-1 in human monocyte and macrophage. Clinical and experimental medi- cine, 15, 245-253.
467. Kim, C. H., Mitchell, J. B., Bursill, C. A., Sowers, A. L., Thet-ford, A., Cook, J. A., ... & Davies, M. J. (2015). The nitroxide radical TEMPOL prevents obesity, hyperlipidaemia, eleva- tion of inflammatory cytokines, and modulates atherosclerotic plaque composition in apoE−/− mice. Atherosclerosis, 240(1), 234-241.
468. Saitoh, T., Kishida, H., Tsukada, Y., Fukuma, Y., Sano, J., Yas- utake, M., ... & Hayakawa, H. (2000). Clinical significance of increased plasma concentration of macrophage colony–stim- ulating factor in patients with angina pectoris. Journal of the American College of Cardiology, 35(3), 655-665.
469. Cybulsky, M. I., Cheong, C., & Robbins, C. S. (2016). Mac- rophages and dendritic cells: partners in atherogenesis. Circu- lation research, 118(4), 637-652.
470. Seshiah, P. N., Kereiakes, D. J., Vasudevan, S. S., Lopes,N., Su, B. Y., Flavahan, N. A., & Goldschmidt-Clermont, P.
J. (2002). Activated monocytes induce smooth muscle cell death: role of macrophage colony-stimulating factor and cell contact. Circulation, 105(2), 174-180.
471. Lind, L., Siegbahn, A., Lindahl, B., Stenemo, M., Sundström, J., & Ärnlöv, J. (2015). Discovery of new risk markers for is- chemic stroke using a novel targeted proteomics chip. Stroke, 46(12), 3340-3347.
472. Andres, V., Pello, O. M., & Silvestre-Roig, C. (2012). Mac- rophage proliferation and apoptosis in atherosclerosis. Cur- rent opinion in lipidology, 23(5), 429-438.
473. Kan, X. H., Zhong, X. Z., Zhang, W. D., & Shi, C. Y. (2016). Increased circulating macrophage-colony stimulating factor and monocyte chemoattractant protein-1 are predictors of in-hospital events in Chinese patients with unstable angina pectoris. Int J Clin Exp Pathol, 9(2), 2021-2026.
474. Branen, L., Hovgaard, L., Nitulescu, M., Bengtsson, E., Nils- son, J., & Jovinge, S. (2004). Inhibition of tumor necrosis factor-α reduces atherosclerosis in apolipoprotein E knock- out mice. Arteriosclerosis, thrombosis, and vascular biology, 24(11), 2137-2142.
475. Kober, F., Canault, M., Peiretti, F., Mueller, C., Kopp, F., Alessi, M. C., ... & Bernard, M. (2007). MRI follow-up of TNF-dependent differential progression of atherosclerotic wall-thickening in mouse aortic arch from early to advanced stages. Atherosclerosis, 195(2), e93-e99.
476. Chew, M., Zhou, J., Daugherty, A., Eriksson, T., Eller- mann-Eriksen, S., Hansen, P. R., & Falk, E. (2003). Thalid- omide inhibits early atherogenesis in apoE-deficient mice. APMIS. Supplementum, (109), 113-116.
477. Boesten, L. S., Zadelaar, A. S. M., van Nieuwkoop, A., Gij- bels, M. J., de Winther, M. P., Havekes, L. M., & van Vlijmen,
B. J. (2005). Tumor necrosis factor-α promotes atherosclerotic lesion progression in APOE* 3-leiden transgenic mice. Cardi- ovascular research, 66(1), 179-185.
478. Canault, M., Peiretti, F., Mueller, C., Kopp, F., Morange, P., Rihs, S., ... & Nalbone, G. (2004). Exclusive expression of transmembrane TNF-α in mice reduces the inflammatory re- sponse in early lipid lesions of aortic sinus. Atherosclerosis, 172(2), 211-218.
479. Prasad, S., Tyagi, A. K., & Aggarwal, B. B. (2016). Detection of inflammatory biomarkers in saliva and urine: potential in diagnosis, prevention, and treatment for chronic diseases. Ex- perimental Biology and Medicine, 241(8), 783-799.
480. WS, W. (2000). C-reactive protein, inflammation and ather- osclerosis: do we really understand it. Eur Heart J, 21, 958- 960.
481. Libby, P., & Ridker, P. M. (2004). Inflammation and athero- sclerosis: role of C-reactive protein in risk assessment. The American journal of medicine, 116(6), 9-16.
482. Ikonomidis, I., Lekakis, J., Revela, I., Andreotti, F., & Ni- hoyannopoulos, P. (2005). Increased circulating C-reactive protein and macrophage-colony stimulating factor are com- plementary predictors of long-term outcome in patients with chronic coronary artery disease. European heart journal, 26(16), 1618-1624.
483. Koenig, W. (2013). High-sensitivity C-reactive protein and atherosclerotic disease: from improved risk prediction to risk-guided therapy. International journal of cardiology, 168(6), 5126-5134.
484. Yu, Q., Liu, Z., Waqar, A. B., Ning, B., Yang, X., Shiomi, M., ... & Fan, J. (2014). Effects of Antisense Oligonucleo- tides against Câ?Reactive Protein on the Development of Ath- erosclerosis in WHHL Rabbits. Mediators of inflammation, 2014(1), 979132.
485. Cossette, E., Cloutier, I., Tardif, K., DonPierre, G., & Tan- guay, J. F. (2013). Estradiol inhibits vascular endothelial cells pro-inflammatory activation induced by C-reactive protein. Molecular and Cellular Biochemistry, 373, 137-147.
486. Wang, Q., Huo, L., He, J., Ding, W., Su, H., Tian, D., ... & Zhu, Y. (2015). Soluble epoxide hydrolase is involved in the development of atherosclerosis and arterial neointima forma- tion by regulating smooth muscle cell migration. American Journal of Physiology-Heart and Circulatory Physiology, 309(11), H1894-H1903.
487. Wu, M. D., Atkinson, T. M., & Lindner, J. R. (2017). Platelets and von Willebrand factor in atherogenesis. Blood, The Jour- nal of the American Society of Hematology, 129(11), 1415- 1419.
488. Ricci, C., & Ferri, N. (2015). Naturally occurring PDGF re- ceptor inhibitors with potential anti-atherosclerotic properties. Vascular pharmacology, 70, 1-7.
489. Lee, M. H., Kwon, B. J., Koo, M. A., You, K. E., & Park, J.C. (2013). Mitogenesis of Vascular Smooth Muscle Cell Stim- ulated by Plateletâ?Derived Growth Factorâ?bb Is Inhibited by Blocking of Intracellular Signaling by Epigallocatechinâ?3â?Oâ? Gallate. Oxidative medicine and cellular longevity, 2013(1), 827905.
490. Sihvola, R. (2003). Platelet-derived growth factor and proin- flammatory cytokines in cardiac allograft arteriosclerosis (Doctoral dissertation, Helsingin yliopisto).
491. Pope III, C. A., Bhatnagar, A., McCracken, J. P., Abplanalp, W., Conklin, D. J., & O’Toole, T. (2016). Exposure to fine particulate air pollution is associated with endothelial injury and systemic inflammation. Circulation research, 119(11), 1204-1214.
492. Chi, H., Messas, E., Levine, R. A., Graves, D. T., & Amar, S. (2004). Interleukin-1 receptor signaling mediates atheroscle- rosis associated with bacterial exposure and/or a high-fat diet in a murine apolipoprotein E heterozygote model: pharmaco- therapeutic implications. Circulation, 110(12), 1678-1685.
493. Von der Thüsen, J. H., Kuiper, J., Van Berkel, T. J., & Biessen,E. A. (2003). Interleukins in atherosclerosis: molecular path- ways and therapeutic potential. Pharmacological reviews, 55(1), 133-166.
494. Vicenová, B., Vopálenský, V., Burýšek, L., & Pospíšek, M. (2009). Emerging role of interleukin-1 in cardiovascular dis- eases. Physiological research, 58(4).
495. Folco, E. J., Sukhova, G. K., Quillard, T., & Libby, P. (2014). Moderate hypoxia potentiates interleukin-1β production in activated human macrophages. Circulation research, 115(10), 875-883.
496. Pagano, P. J., & Gutterman, D. D. (2007). The adventitia: the outs and ins of vascular disease. Cardiovascular research, 75(4), 636-639.
497. Edsfeldt, A., Grufman, H., Asciutto, G., Nitulescu, M., Pers- son, A., Nilsson, M., ... & Gonçalves, I. (2015). Circulating cy- tokines reflect the expression of pro-inflammatory cytokines in atherosclerotic plaques. Atherosclerosis, 241(2), 443-449.
498. Nakai, Y., Iwabuchi, K., Fujii, S., Ishimori, N., Dashtsoodol, N., Watano, K., ... & Onoé, K. (2004). Natural killer T cells accelerate atherogenesis in mice. Blood, 104(7), 2051-2059.
499. Aslanian, A. M., Chapman, H. A., & Charo, I. F. (2005). Tran- sient role for CD1d-restricted natural killer T cells in the for- mation of atherosclerotic lesions. Arteriosclerosis, thrombo- sis, and vascular biology, 25(3), 628-632.
500. Voloshyna, I., Littlefield, M. J., & Reiss, A. B. (2014). Ather- osclerosis and interferon-γ: new insights and therapeutic tar- gets. Trends in cardiovascular medicine, 24(1), 45-51.
501. Harvey, E. J., & Ramji, D. P. (2005). Interferon-γ and athero-sclerosis: pro-or anti-atherogenic?. Cardiovascular research, 67(1), 11-20.
502. Tavakoli, N. N., Harris, A. K., Sullivan, D. R., Hambly, B. D., & Bao, S. (2012). Interferon-γ deficiency reduces neointimal formation in a model of endoluminal endothelial injury com- bined with atherogenic diet. International Journal of Molecu- lar Medicine, 30(3), 545-552.
503. Kunjathoor, V. V., Febbraio, M., Podrez, E. A., Moore, K. J., Andersson, L., Koehn, S., ... & Freeman, M. W. (2002). Scavenger receptors class AI/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. Journal of Biological Chemistry, 277(51), 49982-49988.
504. Febbraio, M., Podrez, E. A., Smith, J. D., Hajjar, D. P., Ha-zen, S. L., Hoff, H. F., ... & Silverstein, R. L. (2000). Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. The Jour- nal of clinical investigation, 105(8), 1049-1056.
505. Rigotti, A. (2000). Scavenger receptors and atherosclerosis.Biological Research, 33(2), 97-103.
506. Bowling, F. L., Rashid, S. T., & Boulton, A. J. (2015). Pre- venting and treating foot complications associated with diabe- tes mellitus. Nature Reviews Endocrinology, 11(10), 606-616.
507. Takeshita, S., Zheng, L. P., Brogi, E., Kearney, M., Pu, L. Q., Bunting, S., ... & Isner, J. M. (1994). Therapeutic angiogene- sis. A single intraarterial bolus of vascular endothelial growth factor augments revascularization in a rabbit ischemic hind limb model. The Journal of clinical investigation, 93(2), 662- 670.
508. Freedman, S. B., & Isner, J. M. (2002). Therapeutic angiogen- esis for coronary artery disease. Annals of internal medicine, 136(1), 54-71.
509. Gupta, R., Tongers, J., & Losordo, D. W. (2009). Human stud- ies of angiogenic gene therapy. Circulation research, 105(8), 724-736.
510. Simons, M., Annex, B. H., Laham, R. J., Kleiman, N., Henry, T., Dauerman, H., ... & Chronos, N. A. (2002). Pharmacolog- ical treatment of coronary artery disease with recombinant fibroblast growth factor-2: double-blind, randomized, con- trolled clinical trial. Circulation, 105(7), 788-793.
511. Karvinen, H., & YlaÌ?-Herttuala, S. (2010). New aspects in vas- cular gene therapy. Current opinion in pharmacology, 10(2), 208-211.
512. Simons, M., & Ware, J. A. (2003). Therapeutic angiogenesis in cardiovascular disease. Nature reviews Drug discovery, 2(11), 863-872.
513. Henry, T. D., Annex, B. H., McKendall, G. R., Azrin, M. A.,Lopez, J. J., Giordano, F. J., ... & McCluskey, E. R. (2003). The VIVA trial: Vascular endothelial growth factor in Ischemia for Vascular Angiogenesis. Circulation, 107(10), 1359-1365.
514. Björkegren, J. L., & Lusis, A. J. (2022). Atherosclerosis: re- cent developments. Cell, 185(10), 1630-1645.
515. Herrmann, J., Lerman, L. O., Rodriguez-Porcel, M., Holmes Jr, D. R., Richardson, D. M., Ritman, E. L., & Lerman, A. (2001). Coronary vasavasorum neovascularization precedes epicardial endothelial dysfunction in experimental hypercho-lesterolemia. Cardiovascular research, 51(4), 762-766.
516. Kirichenko, A. K., Shulmin, A. V., Sharkova, A. F., Patlataya,N. N., & Bolshakov, I. N. (2017). Morphological reconstruc- tion of main arteries by perivascular implantation of sulfat- ed chitosan in experimental atherosclerosis. СовÑ?еменнÑ?е Ñ?еÑ?нологии в медиÑ?ине, 9(4 (eng)), 115-120.
517. Polignano, R., Baggiore, C., Falciani, F., Restelli, U., Troi- si, N., Michelagnoli, S., ... & Landini, G. (2016). Efficacy, safety and feasibility of intravenous iloprost in the domiciliary treatment of patients with ischemic disease of the lower limbs. European Review for Medical & Pharmacological Sciences, 20(17).
518. Martel, C., Li, W., Fulp, B., Platt, A. M., Gautier, E. L., West- erterp, M., ... & Randolph, G. J. (2013). Lymphatic vasculature mediates macrophage reverse cholesterol transport in mice. The Journal of clinical investigation, 123(4), 1571-1579.
519. Langheinrich, A. C., Michniewicz, A., Bohle, R. M., & Rit- man, E. L. (2007). Vasa vasorum neovascularization and le- sion distribution among different vascular beds in ApoE−/−/ LDL−/− double knockout mice. Atherosclerosis, 191(1), 73- 81.
520. Milasan, A., Dallaire, F., Mayer, G., & Martel, C. (2016). Ef- fects of LDL receptor modulation on lymphatic function. Sci- entific reports, 6(1), 27862.
521. Drozdz, K., Janczak, D., Dziegiel, P., Podhorska, M., Pi- otrowska, A., Patrzalek, D., ... & Szuba, A. (2012). Adventitial lymphatics and atherosclerosis. Lymphology, 45(1), 26-33.
522. Kholová, I., Dragneva, G., Ä?ermáková, P., Laidinen, S., KaskenpaÌ?aÌ?, N., Hazes, T., ... & YlaÌ?â?Herttuala, S. (2011). Lym- phatic vasculature is increased in heart valves, ischaemic and inflamed hearts and in cholesterolâ?rich and calcified athero- sclerotic lesions. European journal of clinical investigation, 41(5), 487-497.
523. Kim, H., Kataru, R. P., & Koh, G. Y. (2012). Regulation and implications of inflammatory lymphangiogenesis. Trends in immunology, 33(7), 350-356.
524. Murdoch, C., Muthana, M., Coffelt, S. B., & Lewis, C. E. (2008). The role of myeloid cells in the promotion of tumour angiogenesis. Nature reviews cancer, 8(8), 618-631.
525. Grunewald, M., Avraham, I., Dor, Y., Bachar-Lustig, E., Itin, A., Yung, S., ... & Keshet, E. (2006). VEGF-induced adult ne- ovascularization: recruitment, retention, and role of accessory cells. Cell, 124(1), 175-189.
526. Dimova, I., Karthik, S., Makanya, A., Hlushchuk, R., Semela, D., Volarevic, V., & Djonov, V. (2019). SDF 1/CXCR4 sig- nalling is involved in blood vessel growth and remodelling by intussusception. Journal of cellular and molecular medicine, 23(6), 3916-3926.
527. Dimova, I., Karthik, S., Makanya, A., Hlushchuk, R., Semela, D., Volarevic, V., & Djonov, V. (2019). SDFâ?1/CXCR4 sig- nalling is involved in blood vessel growth and remodelling by intussusception. Journal of cellular and molecular medicine, 23(6), 3916-3926.
528. Ochoa, C. D., Wu, R. F., & Terada, L. S. (2018). ROS sign- aling and ER stress in cardiovascular disease. Molecular as- pects of medicine, 63, 18-29.
529. Alencar, G. F., Owsiany, K. M., Karnewar, S., Sukhavasi, K., Mocci, G., Nguyen, A. T., ... & Owens, G. K. (2020). Stem cell pluripotency genes Klf4 and Oct4 regulate complex SMC phenotypic changes critical in late-stage atherosclerotic lesion pathogenesis. Circulation, 142(21), 2045-2059.
530. Pan, H., Xue, C., Auerbach, B. J., Fan, J., Bashore, A. C., Cui, J., ... & Reilly, M. P. (2020). Single-cell genomics reveals a novel cell state during smooth muscle cell phenotypic switch- ing and potential therapeutic targets for atherosclerosis in mouse and human. Circulation, 142(21), 2060-2075.
531. Lacolley, P., Regnault, V., Nicoletti, A., Li, Z., & Michel, J. B. (2012). The vascular smooth muscle cell in arterial patholo- gy: a cell that can take on multiple roles. Cardiovascular re- search, 95(2), 194-204.
532. He, C., Hu, X., Weston, T. A., Jung, R. S., Sandhu, J., Huang, S., ... & Young, S. G. (2018). Macrophages release plasma membrane-derived particles rich in accessible cholesterol. Proceedings of the National Academy of Sciences, 115(36), E8499-E8508.
533. Butoi, E. D., Gan, A. M., Manduteanu, I., Stan, D., Calin, M., Pirvulescu, M., ... & Simionescu, M. (2011). Cross talk be- tween smooth muscle cells and monocytes/activated mono- cytes via CX3CL1/CX3CR1 axis augments expression of pro-atherogenic molecules. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research, 1813(12), 2026-2035.
534. Gan, A. M., Pirvulescu, M. M., Stan, D., Simion, V., Calin, M., Manduteanu, I., & Butoi, E. (2013). Monocytes and smooth muscle cells crossâ?talk activates STAT3 and induces resistin and reactive oxygen species and production. Journal of cellular biochemistry, 114(10), 2273-2283.
535. Vijayagopal, P., & Glancy, D. L. (1996). Macrophages stimu- late cholesteryl ester accumulation in cocultured smooth mus- cle cells incubated with lipoprotein-proteoglycan complex. Arteriosclerosis, thrombosis, and vascular biology, 16(9), 1112-1121.
536. Weinert, S., Poitz, D. M., Auffermann-Gretzinger, S., Eger, L., Herold, J., Medunjanin, S., ... & Braun-Dullaeus, R. C. (2013). The lysosomal transfer of LDL/cholesterol from mac- rophages into vascular smooth muscle cells induces their phe- notypic alteration. Cardiovascular research, 97(3), 544-552.
537. Newby, A. C., & Zaltsman, A. B. (1999). Fibrous cap for- mation or destruction—the critical importance of vascular smooth muscle cell proliferation, migration and matrix for- mation. Cardiovascular research, 41(2), 345-360.
538. Lu, Q., Yao, Y., Hu, Z., Hu, C., Song, Q., Ye, J., ... & Wang,Q. K. (2016). Angiogenic factor AGGF1 activates autophagy with an essential role in therapeutic angiogenesis for heart dis- ease. PLoS Biology, 14(8), e1002529.
539. Zhang, J., Kasim, V., Xie, Y. D., Huang, C., Sisjayawan, J., Dwi Ariyanti, A., ... & Wu, S. R. (2017). Inhibition of PHD3 by salidroside promotes neovascularization through cell–cell communications mediated by muscle-secreted angiogenic factors. Scientific reports, 7(1), 43935.
540. Luo, L. L., Han, J. X., Wu, S. R., & Kasim, V. (2022). In-tramuscular injection of sotagliflozin promotes neovascular- ization in diabetic mice through enhancing skeletal muscle cells paracrine function. Acta Pharmacologica Sinica, 43(10), 2636-2650.
541. Liu, C., Han, J., Marcelina, O., Nugrahaningrum, D. A., Huang, S., Zou, M., ... & Kasim, V. (2021). Discovery of sal- idroside-derivated glycoside analogues as novel angiogenesis agents to treat diabetic hind limb ischemia. Journal of Medic- inal Chemistry, 65(1), 135-162.
542. Fallah, A., Sadeghinia, A., Kahroba, H., Samadi, A., Heidari,H. R., Bradaran, B., ... & Molavi, O. (2019). Therapeutic targeting of angiogenesis molecular pathways in angiogen- esis-dependent diseases. Biomedicine & Pharmacotherapy, 110, 775-785.
543. Folkman, J. (2004). Endogenous angiogenesis inhibitors. Ap- mis, 112(7â?8), 496-507.
544. Vasilyeva, N. Y., Levdanskya, A. V., Kuznetsova, B. N., Skvortsova, G. P., Kazachenko, A. S., Djakovitch, L., Pinel,
C. (2016). Synthesis of Sulfated Arabinogalactan Derivatives with Histidine and Arginine. Journal of Siberian Federal Uni- versity Chemistry, 9(3), 318-325
545. Quiñones, J. P., Peniche, H., & Peniche, C. (2018). Chitosan based self-assembled nanoparticles in drug delivery. Poly- mers, 10(3), 235.
546. Cheng, Y., Xie, Y., Shi, L., Xing, Y., Guo, S., Gao, Y., ... & Shi,B. (2022). Effects of rare earth-chitosan chelate on growth performance, antioxidative and immune function in broilers. Italian Journal of Animal Science, 21(1), 303-313.
547. Imam, S. S., Alshehri, S., Altamimi, M. A., Almalki, R. K. H.,
Hussain, A., Bukhari, S. I., ... & Qamar, W. (2022). Formula- tion of chitosan-coated apigenin bilosomes: in vitro character- ization, antimicrobial and cytotoxicity assessment. Polymers, 14(5), 921.
548. Yu, J., Ruan, Q., Nie, X., Yu, L., & Huang, B. (2020). Syn-thetic CD47 antibody-chitosan/hyaluronic acid polyelectro- lyte complex mediates targeted inhibition of atherosclerotic plaques by exogenous foam-like cells via the NLRP3 pathway. Journal of Biomaterials Applications, 34(10), 1381-1394.
549. Nguyen, M. A., Wyatt, H., Susser, L., Geoffrion, M., Rash- eed, A., Duchez, A. C., ... & Rayner, K. J. (2019). Delivery of microRNAs by chitosan nanoparticles to functionally alter macrophage cholesterol efflux in vitro and in vivo. ACS nano, 13(6), 6491-6505.
550. Yu, J., Ruan, Q., Nie, X., Yu, L., & Huang, B. (2020). Syn-
thetic CD47 antibody-chitosan/hyaluronic acid polyelectro- lyte complex mediates targeted inhibition of atherosclerotic plaques by exogenous foam-like cells via the NLRP3 pathway. Journal of Biomaterials Applications, 34(10), 1381-1394.
551. Liu, Y., Liu, D., Zhu, L., Gan, Q., & Le, X. (2015). Temperature-dependent structure stability and in vitro release of chi- tosan-coated curcumin liposome. Food research internation- al, 74, 97-105.
552. Talbot, C. P., Plat, J., Ritsch, A., & Mensink, R. P. (2018). De- terminants of cholesterol efflux capacity in humans. Progress in lipid research, 69, 21-32.
553. Van Gils, J. M., Derby, M. C., Fernandes, L. R., Ramkhel- awon, B., Ray, T. D., Rayner, K. J., ... & Moore, K. J. (2012). The neuroimmune guidance cue netrin-1 promotes athero-sclerosis by inhibiting the emigration of macrophages from plaques. Nature immunology, 13(2), 136-143.
554. Wenli, D., Zirong, X., Xinyan, H., Yinglei, X., & Zhiguo, M. (2008). Preparation, characterization and adsorption proper- ties of chitosan nanoparticles for eosin Y as a model anionic dye.
555. Rice, J. J., Martino, M. M., De Laporte, L., Tortelli, F., Bri- quez, P. S., & Hubbell, J. A. (2013). Engineering the regener- ative microenvironment with biomaterials. Advanced health- care materials, 2(1), 57-71.
556. Moulisová, V., Gonzalez-García, C., Cantini, M., Rod- rigo-Navarro, A., Weaver, J., Costell, M., ... & Salm- erón-Sánchez, M. (2017). Engineered microenvironments for synergistic VEGF–Integrin signalling during vascularization. Biomaterials, 126, 61-74.
557. Mitragotri, S., & Lahann, J. (2009). Physical approaches to biomaterial design. Nature materials, 8(1), 15-23.
558. Laham, R. J., Sellke, F. W., Edelman, E. R., Pearlman, J. D.,Ware, J. A., Brown, D. L., ... & Simons, M. (1999). Local perivascular delivery of basic fibroblast growth factor in pa- tients undergoing coronary bypass surgery: results of a phase I randomized, double-blind, placebo-controlled trial. Circula- tion, 100(18), 1865-1871.
559. Ruel, M., Laham, R. J., Parker, J. A., Post, M. J., Ware, J. A., Simons, M., & Sellke, F. W. (2002). Long-term effects of surgical angiogenic therapy with fibroblast growth factor 2 protein. The Journal of thoracic and cardiovascular surgery, 124(1), 28-34.
560. Barc, P., Plonek, T., Baczynska, D., Radwanska, A., Witk- iewicz, W., Halon, A., ... & Skora, J. (2016). A combination of VEGF165/HGF genes is more effective in blood vessels formation than ANGPT1/VEGF165 genes in an in vivo rat model. Int J Clin Exp Med, 9(7), 12737-12744.
561. Makarevich, P. I., Boldyreva, M. A., Gluhanyuk, E. V., Efi- menko, A. Y., Dergilev, K. V., Shevchenko, E. K., ... & Parfyonova, Y. V. (2015). Enhanced angiogenesis in ischemic skeletal muscle after transplantation of cell sheets from bac- ulovirus-transduced adipose-derived stromal cells expressing VEGF165. Stem cell research & therapy, 6, 1-20.
562. Vemulapalli, S., Patel, M. R., & Jones, W. S. (2015). Limb is- chemia: cardiovascular diagnosis and management from head to toe. Current cardiology reports, 17, 1-12.
563. Cooke, J. P., & Losordo, D. W. (2015). Modulating the vascu- lar response to limb ischemia: angiogenic and cell therapies. Circulation research, 116(9), 1561-1578.
564. Lederman, R. J., Mendelsohn, F. O., Anderson, R. D., Sauce- do, J. F., Tenaglia, A. N., Hermiller, J. B., ... & Annex, B. H. (2002). Therapeutic angiogenesis with recombinant fibroblast growth factor-2 for intermittent claudication (the TRAFFIC study): a randomised trial. The Lancet, 359(9323), 2053-2058.
565. Jazwa, A., Florczyk, U., Grochot-Przeczek, A., Krist, B., Lo- boda, A., Jozkowicz, A., & Dulak, J. (2016). Limb ischemia and vessel regeneration: Is there a role for VEGF?. Vascular pharmacology, 86, 18-30.
566. Curry, C. W., Sturgeon, S. M., O’Grady, B. J., Yates, A., Kjar, A., Paige, H., ... & Lippmann, E. S. (2023). Growth factor free, peptide-functionalized gelatin hydrogel promotes arteri- ogenesis and attenuates tissue damage in a murine model of critical limb ischemia. Biomaterials, 303, 122397.
567. Patel, Z. S., & Mikos, A. G. (2004). Angiogenesis with bio- material-based drug-and cell-delivery systems. Journal of Bi- omaterials Science, Polymer Edition, 15(6), 701-726.
568. King, W. J., & Krebsbach, P. H. (2012). Growth factor deliv- ery: how surface interactions modulate release in vitro and in vivo. Advanced drug delivery reviews, 64(12), 1239-1256.
569. Nicosia, A., Salamone, M., Costa, S., Ragusa, M. A., & Gher- si, G. (2023). Mimicking molecular pathways in the design of smart hydrogels for the design of vascularized engineered tissues. International Journal of Molecular Sciences, 24(15), 12314.
570. Stamati, K., Priestley, J. V., Mudera, V., & Cheema, U. (2014). Laminin promotes vascular network formation in 3D in vitro collagen scaffolds by regulating VEGF uptake. Experimental cell research, 327(1), 68-77.
571. Haggerty, A. E., Maldonado-Lasunción, I., & Oudega, M. (2018). Biomaterials for revascularization and immunomod- ulation after spinal cord injury. Biomedical materials, 13(4), 044105.
572. Song, H. H. G., Rumma, R. T., Ozaki, C. K., Edelman, E. R., & Chen, C. S. (2018). Vascular tissue engineering: progress, challenges, and clinical promise. Cell stem cell, 22(3), 340- 354.
573. Aravamudhan, A., M. Ramos, D., Nip, J., Subramanian, A., James, R., D. Harmon, M., ... & G. Kumbar, S. (2013). Os- teoinductive small molecules: growth factor alternatives for bone tissue engineering. Current pharmaceutical design, 19(19), 3420-3428.
574. Simón-Yarza, T., Formiga, F. R., Tamayo, E., Pelacho, B., Prosper, F., & Blanco-Prieto, M. J. (2012). Vascular endothe- lial growth factor-delivery systems for cardiac repair: an over- view. Theranostics, 2(6), 541.
575. Kumagai, M., Marui, A., Tabata, Y., Takeda, T., Yamamoto, M., Yonezawa, A., ... & Sakata, R. (2016). Safety and effica- cy of sustained release of basic fibroblast growth factor using gelatin hydrogel in patients with critical limb ischemia. Heart and vessels, 31, 713-721.
576. Foster, G. A., Headen, D. M., González-García, C., Salm- erón-Sánchez, M., Shirwan, H., & García, A. J. (2017). Pro- tease-degradable microgels for protein delivery for vasculari- zation. Biomaterials, 113, 170-175.
577. Yao, S., Yang, Y., Wang, X., & Wang, L. (2017). Fabrication and characterization of aligned fibrin nanofiber hydrogel loaded with PLGA microspheres. Macromolecular Research, 25(6), 528-533.
578. Zhang, J., Xia, W., Liu, P., Cheng, Q., Tahirou, T., Gu, W., & Li, B. (2010). Chitosan modification and pharmaceutical/bio- medical applications. Marine drugs, 8(7), 1962-1987.
579. Fischbach, C., & Mooney, D. J. (2007). Polymers for pro-and anti-angiogenic therapy. Biomaterials, 28(12), 2069-2076.
580. Murphy, W. L., Peters, M. C., Kohn, D. H., & Mooney, D. J. (2000). Sustained release of vascular endothelial growth fac- tor from mineralized poly (lactide-co-glycolide) scaffolds for tissue engineering. Biomaterials, 21(24), 2521-2527.
581. Wang, Z., Wang, Z., Lu, W. W., Zhen, W., Yang, D., & Peng, S. (2017). Novel biomaterial strategies for controlled growth factor delivery for biomedical applications. NPG Asia Mate- rials, 9(10), e435-e435.
582. Ziegler, J., Anger, D., Krummenauer, F., Breitig, D., Fickert, S., & Guenther, K. P. (2008). Biological activity of recombi- nant human growth factors released from biocompatible bone implants. Journal of Biomedical Materials Research Part A: An Oficial Journal of The Society for Biomaterials, The Jap- anese Society for Biomaterials, and The Australian Society for Biomaterials and the Korean Society for Biomaterials, 86(1), 89-97.
583. Ehrbar, M., Schoenmakers, R., Christen, E. H., Fusseneg- ger, M., & Weber, W. (2008). Drug-sensing hydrogels for the inducible release of biopharmaceuticals. Nature materials, 7(10), 800-804.
584. Masters, K. S. (2011). Covalent growth factor immobilization strategies for tissue repair and regeneration. Macromolecular bioscience, 11(9), 1149-1163.
585. Martino, M. M., Briquez, P. S., Maruyama, K., & Hubbell, J.A. (2015). Extracellular matrix-inspired growth factor deliv- ery systems for bone regeneration. Advanced drug delivery reviews, 94, 41-52.
586. Naito, Y., Shinoka, T., Duncan, D., Hibino, N., Solomon, D., Cleary, M., ... & Breuer, C. (2011). Vascular tissue engineer- ing: towards the next generation vascular grafts. Advanced drug delivery reviews, 63(4-5), 312-323.
587. Freeman, I., & Cohen, S. (2009). The influence of the sequen- tial delivery of angiogenic factors from affinity-binding algi- nate scaffolds on vascularization. Biomaterials, 30(11), 2122- 2131.
588. Freeman, I., Kedem, A., & Cohen, S. (2008). The effect of sulfation of alginate hydrogels on the specific binding and controlled release of heparin-binding proteins. Biomaterials, 29(22), 3260-3268.
589. Phadke, G., Hanna, R. M., Ferrey, A., Torres, E. A., Singla, A., Kaushal, A., ... & Jhaveri, K. D. (2021). Review of intra- vitreal VEGF inhibitor toxicity and report of collapsing FSGS with TMA in a patient with age-related macular degeneration. Clinical Kidney Journal, 14(10), 2158-2165.
590. Butt, O. I., Carruth, R., Kutala, V. K., Kuppusamy, P., & Mol- dovan, N. I. (2007). Stimulation of peri-implant vasculariza- tion with bone marrow-derived progenitor cells: monitoring by in vivo EPR oximetry. Tissue engineering, 13(8), 2053- 2061.
591. Wei, Z., Schnellmann, R., Pruitt, H. C., & Gerecht, S. (2020). Hydrogel network dynamics regulate vascular morphogene- sis. Cell stem cell, 27(5), 798-812.
592. Singh, S., Wu, B. M., & Dunn, J. C. (2011). The enhancement of VEGF-mediated angiogenesis by polycaprolactone scaf- folds with surface cross-linked heparin. Biomaterials, 32(8), 2059-2069.
593. Levenberg, S., Rouwkema, J., Macdonald, M., Garfein, E. S., Kohane, D. S., Darland, D. C., ... & Langer, R. (2005). Engi- neering vascularized skeletal muscle tissue. Nature biotechnology, 23(7), 879-884.
594. Chen, Y. C., Lin, R. Z., Qi, H., Yang, Y., Bae, H., Melero Martin, J. M., & Khademhosseini, A. (2012). Functional hu- man vascular network generated in photocrosslinkable gela- tin methacrylate hydrogels. Advanced functional materials, 22(10), 2027-2039.
595. Santos, M. I., Fuchs, S., Gomes, M. E., Unger, R. E., Reis, R. L., & Kirkpatrick, C. J. (2007). Response of micro-and mac- rovascular endothelial cells to starch-based fiber meshes for bone tissue engineering. Biomaterials, 28(2), 240-248.
596. Unger, R. E., Dohle, E., & Kirkpatrick, C. J. (2015). Improv- ing vascularization of engineered bone through the generation of pro-angiogenic effects in co-culture systems. Advanced drug delivery reviews, 94, 116-125.
597. Banno, K., & Yoder, M. C. (2018). Tissue regeneration using endothelial colony-forming cells: promising cells for vascular repair. Pediatric research, 83(1), 283-290.
598. Perets, A., Baruch, Y., Weisbuch, F., Shoshany, G., Neufeld, G., & Cohen, S. (2003). Enhancing the vascularization of threeâ?dimensional porous alginate scaffolds by incorporating controlled release basic fibroblast growth factor microspheres. Journal of Biomedical Materials Research Part A: An Oficial Journal of The Society for Biomaterials, The Japanese Society for Biomaterials, and The Australian Society for Biomaterials and the Korean Society for Biomaterials, 65(4), 489-497.
599. Van Rensburg, A. J., Davies, N. H., Oosthuysen, A., Choko- za, C., Zilla, P., & Bezuidenhout, D. (2017). Improved vascu- larization of porous scaffolds through growth factor delivery from heparinized polyethylene glycol hydrogels. Acta bioma- terialia, 49, 89-100.
600. Newman, A. C., Nakatsu, M. N., Chou, W., Gershon, P. D., & Hughes, C. C. (2011). The requirement for fibroblasts in angiogenesis: fibroblast-derived matrix proteins are essential for endothelial cell lumen formation. Molecular biology of the cell, 22(20), 3791-3800.
601. Berthod, F., Symes, J., Tremblay, N., Medin, J. A., & Auger,
F. A. (2012). Spontaneous fibroblastâ?derived pericyte recruit- ment in a human tissueâ?engineered angiogenesis model in vit- ro. Journal of cellular physiology, 227(5), 2130-2137.
602. Abdul Sisak, M. A., Louis, F., & Matsusaki, M. (2020). In vitro fabrication and application of engineered vascular hy- drogels. Polymer Journal, 52(8), 871-881.
603. Kaplanskaya, I. B., YeN, G., & Frank, G. A. (2005). Angio- genesis, intercellular contacts and stromal-parenchymal rela- tionships in norm and pathology. Russian Journal of Oncol- ogy, 4.
604. Deepa, R., Paul, W., Anilkumar, T. V., & Sharma, C. P. (2013). Differential healing of full thickness rabbit skin wound by fi- broblast loaded chitosan sponge. Journal of Biomaterials and Tissue Engineering, 3(3), 261-272.
605. Sun, H., Wang, X., Hu, X., Yu, W., You, C., Hu, H., & Han,
C. (2012). Promotion of angiogenesis by sustained release of rhGMâ?CSF from heparinized collagen/chitosan scaffolds. Journal of Biomedical Materials Research Part B: Applied Biomaterials, 100(3), 788-798.
606. Wang, P., Liu, J., & Zhang, T. (2013). In vitro biocompatibility of electrospun chitosan/collagen scaffold. Journal of Nano- materials, 2013(1), 958172.
607. Sarrazin, S., Lamanna, W. C., & Esko, J. D. (2011). Heparan sulfate proteoglycans. Cold Spring Harbor perspectives in bi- ology, 3(7), a004952.
608. Thomas, A. M., Gomez, A. J., Palma, J. L., Yap, W. T., & Shea, L. D. (2014). Heparin–chitosan nanoparticle function- alization of porous poly (ethylene glycol) hydrogels for local- ized lentivirus delivery of angiogenic factors. Biomaterials, 35(30), 8687-8693.
609. Bolshakov, I. N., Shestakova, L. A., Kotikov, A. R., & Kap- tyuk, G. I. (2013). Experimental atherosclerosis in rats. Mor- phological reconstruction of the main artery wall with the pol- ysaccharide biopolymers. Fundam. Res, 10, 557-563.
610. Alavi, M., & Nokhodchi, A. (2020). An overview on antimi- crobial and wound healing properties of ZnO nanobiofilms, hydrogels, and bionanocomposites based on cellulose, chi- tosan, and alginate polymers. Carbohydrate polymers, 227, 115349.
611. Lee, S., Valmikinathan, C. M., Byun, J., Kim, S., Lee, G., Mokarram, N., ... & Yoon, Y. S. (2015). Enhanced therapeutic neovascularization by CD31-expressing cells and embryonic stem cell-derived endothelial cells engineered with chitosan hydrogel containing VEGF-releasing microtubes. Biomateri- als, 63, 158-167.
612. Hasan, A., Khattab, A., Islam, M. A., Hweij, K. A., Zeitouny, J., Waters, R., ... & Paul, A. (2015). Injectable hydrogels for cardiac tissue repair after myocardial infarction. Advanced Science, 2(11), 1500122.
613. Vieira, T., Silva, J. C., Do Rego, A. B., Borges, J. P., & Henriques, C. (2019). Electrospun biodegradable chitosan based-poly (urethane urea) scaffolds for soft tissue engineer- ing. Materials Science and Engineering: C, 103, 109819.
614. Zhao, N., Yue, Z., Cui, J., Yao, Y., Song, X., Cui, B., ... &
Li, Z. (2019). IGF-1C domain-modified hydrogel enhances therapeutic potential of mesenchymal stem cells for hindlimb ischemia. Stem cell research & therapy, 10, 1-12.
615. Rouwkema, J., & Khademhosseini, A. (2016). Vascularization and angiogenesis in tissue engineering: beyond creating static networks. Trends in biotechnology, 34(9), 733-745.
616. Alsop, A. T., Pence, J. C., Weisgerber, D. W., Harley, B. A., & Bailey, R. C. (2014). Photopatterning of vascular endothelial growth factor within collagen-glycosaminoglycan scaffolds can induce a spatially confined response in human umbilical vein endothelial cells. Acta biomaterialia, 10(11), 4715-4722.
617. Baker, B. M., Trappmann, B., Stapleton, S. C., Toro, E., & Chen, C. S. (2013). Microfluidics embedded within extracel- lular matrix to define vascular architectures and pattern diffu- sive gradients. Lab on a Chip, 13(16), 3246-3252.
618. Miller, J. S., Stevens, K. R., Yang, M. T., Baker, B. M., Nguyen,
D. H. T., Cohen, D. M., ... & Chen, C. S. (2012). Rapid cast- ing of patterned vascular networks for perfusable engineered three-dimensional tissues. Nature materials, 11(9), 768-774.
619. Zheng, Y., Chen, J., Craven, M., Choi, N. W., Totorica, S., Di- az-Santana, A., ... & Stroock, A. D. (2012). In vitro microves- sels for the study of angiogenesis and thrombosis. Proceedings of the national academy of sciences, 109(24), 9342-9347. - 620.Kirichenko, Ð. Ð?., Patlataya, N. N., Sharkova, Ð. F., Pevnev, Ð. Ð., Kontorev, Ð?. V., Shapovalova, Ð?. V., ... & Bolshakov,I. N. (2017). Pathomorphism of limb major vessels in exper- imental atherogenic inflammation. The role of adventitial in- timal relations. СовÑ?еменнÑ?е Ñ?еÑ?нологии в медиÑ?ине, 9(3 (eng)), 162-173.
- 621. Bolshakov, I. N., Shestakova, L. A., Kotikov, A. R., Kaptyuk,G. I. (2013). Experimental atherogenic inflammation of the great arteries in rabbits. Minimally invasive technology for morphological reconstruction of the vascular wall in the early stages of atherogenesis. J Fundamental Research.
622. Blatchley, M. R., & Gerecht, S. (2015). Acellular implantable and injectable hydrogels for vascular regeneration. Biomedi- cal Materials, 10(3), 034001.
623. Mongiat, M., Andreuzzi, E., Tarticchio, G., & Paulitti, A. (2016). Extracellular matrix, a hard player in angiogenesis. International journal of molecular sciences, 17(11), 1822.
624. Navab, M., Hough, G. P., Stevenson, L. W., Drinkwater, D. C., Laks, H., & Fogelman, A. M. (1988). Monocyte migration into the subendothelial space of a coculture of adult human aortic endothelial and smooth muscle cells. The Journal of clinical investigation, 82(6), 1853-1863.
625. Murphy, S. V., De Coppi, P., & Atala, A. (2020). Opportu- nities and challenges of translational 3D bioprinting. Nature biomedical engineering, 4(4), 370-380.
626. Yang, J., Dang, H., & Xu, Y. (2022). Recent advancement of decellularization extracellular matrix for tissue engineering and biomedical application. Artificial organs, 46(4), 549-567.
627. Klingenberg, R., & Hansson, G. K. (2009). Treating inflam- mation in atherosclerotic cardiovascular disease: emerging therapies. European heart journal, 30(23), 2838-2844.
628. Ding, X., Gao, J., Wang, Z., Awada, H., & Wang, Y. (2016). A shear-thinning hydrogel that extends in vivo bioactivity of FGF2. Biomaterials, 111, 80-89.
629. Shahzadi, L., Yar, M., Jamal, A., Siddiqi, S. A., Chaudhry, A. A., Zahid, S., ... & MacNeil, S. (2016). Triethyl orthoformate covalently cross-linked chitosan-(poly vinyl) alcohol based biodegradable scaffolds with heparin-binding ability for pro- moting neovascularisation. Journal of biomaterials applica- tions, 31(4), 582-593.
630. Yar, M., Gigliobianco, G., Shahzadi, L., Dew, L., Siddiqi, S. A., Khan, A. F., ... & MacNeil, S. (2016). Production of chi- tosan PVA PCL hydrogels to bind heparin and induce angi- ogenesis. International Journal of Polymeric Materials and Polymeric Biomaterials, 65(9), 466-476.
631. Adrian, E., Treľová, D., Filová, E., Kumorek, M., Lobaz, V., Poreba, R., ... & Kubies, D. (2021). Complexation of CXCL12, FGF-2 and VEGF with heparin modulates the pro- tein release from alginate microbeads. International Journal of Molecular Sciences, 22(21), 11666.
632. Jeon, O., Lee, K., & Alsberg, E. (2018). Spatial micropattern- ing of growth factors in 3D hydrogels for locationâ?specific regulation of cellular behaviors. Small, 14(25), 1800579.
633. Nilasaroya, A., Kop, A. M., & Morrison, D. A. (2021). Hepa- rin functionalized hydrogels as growth factorâ?signaling sub- strates. Journal of Biomedical Materials Research Part A, 109(3), 374-384.
634. Claaßen, C., Sewald, L., Tovar, G. E., & Borchers, K. (2017). Controlled release of vascular endothelial growth factor from heparin-functionalized gelatin type A and albumin hydrogels. Gels, 3(4), 35.
635. Lu, Q., Li, M., Zou, Y., & Cao, T. (2014). Delivery of basic fibroblast growth factors from heparinized decellularized adi- pose tissue stimulates potent de novo adipogenesis. Journal of Controlled Release, 174, 43-50.
636. Sheridan, M. H., Shea, L. D., Peters, M. C., & Mooney, D. J. (2000). Bioabsorbable polymer scaffolds for tissue engineer- ing capable of sustained growth factor delivery. Journal of controlled release, 64(1-3), 91-102.
637. Zhang, J., Li, G., Gao, S., Yao, Y., Pang, L., Li, Y., ... & Li,C. (2014). Monocyte chemoattractant protein-1 released from polycaprolactone/chitosan hybrid membrane to promote an- giogenesis in vivo. Journal of bioactive and compatible poly- mers, 29(6), 572-588.
638. Obara, K., Ishihara, M., Fujita, M., Kanatani, Y., Hattori, H., Matsui, T., ... & Maehara, T. (2005). Acceleration of wound healing in healingâ?impaired db/db mice with a photocrosslink- able chitosan hydrogel containing fibroblast growth factorâ?2. Wound repair and regeneration, 13(4), 390-397.
639. Wang, W., Lin, S., Xiao, Y., Huang, Y., Tan, Y., Cai, L., & Li, X. (2008). Acceleration of diabetic wound healing with chitosan-crosslinked collagen sponge containing recombinant human acidic fibroblast growth factor in healing-impaired STZ diabetic rats. Life sciences, 82(3-4), 190-204.
640. Yoon, J. J., Chung, H. J., Lee, H. J., & Park, T. G. (2006).
Heparinâ?immobilized biodegradable scaffolds for local and sustained release of angiogenic growth factor. Journal of bio- medical materials research Part A, 79(4), 934-942.
641. Wissink, M. J. B., Beernink, R., Poot, A. A., Engbers, G. H.M., Beugeling, T., Van Aken, W. G., & Feijen, J. (2000). Im- proved endothelialization of vascular grafts by local release of growth factor from heparinized collagen matrices. Journal of Controlled Release, 64(1-3), 103-114.
642. Gospodarowicz, D., & Cheng, J. (1986). Heparin protects basic and acidic FGF from inactivation. Journal of cellular physiology, 128(3), 475-484.
643. Zhang, L., Furst, E. M., & Kiick, K. L. (2006). Manipulation of hydrogel assembly and growth factor delivery via the use of peptide–polysaccharide interactions. Journal of controlled release, 114(2), 130-142.
644. Shen, H., Hu, X., Yang, F., Bei, J., & Wang, S. (2011). Cell affinity for bFGF immobilized heparin-containing poly (lac- tide-co-glycolide) scaffolds. Biomaterials, 32(13), 3404-3412.
645. Minardi, S., Pandolfi, L., Taraballi, F., Wang, X., De Rosa, E., Mills, Z. D., ... & Tasciotti, E. (2017). Enhancing vasculariza- tion through the controlled release of platelet-derived growth factor-BB. ACS applied materials & interfaces, 9(17), 14566- 14575.
646. Kwee, B. J., Budina, E., Najibi, A. J., & Mooney, D. J. (2018). CD4 T-cells regulate angiogenesis and myogenesis. Biomate- rials, 178, 109-121.
647. Lee, K. Y., & Mooney, D. J. (2012). Alginate: properties and biomedical applications. Progress in polymer science, 37(1), 106-126.
648. Jain, A., Gupta, Y., & Jain, S. K. (2007). Perspectives of bio- degradable natural polysaccharides for site-specific drug de- livery to the colon. J Pharm Pharm Sci, 10(1), 86-128.
649. Silva, E. A., & Mooney, D. J. (2010). Effects of VEGF tem- poral and spatial presentation on angiogenesis. Biomaterials, 31(6), 1235-1241.
650. Lee, J., Bhang, S. H., Park, H., Kim, B. S., & Lee, K. Y.
(2010). Active blood vessel formation in the ischemic hind- limb mouse model using a microsphere/hydrogel combination system. Pharmaceutical research, 27, 767-774.
651. Gonzalez-Pujana, A., Orive, G., Pedraz, J. L., Santos-Vizcai- no, E., Hernandez, R. M. (2018). Alginates and Their Bio- medical Applications. Springer; Berlin/Heidelberg, Germany, Alginate Microcapsules for Drug Delivery.
652. Aparicio-Collado, J. L., García-San-Martín, N., Molina-Ma- teo, J., Cabanilles, C. T., Quiles, V. D., Serrano-Aroca, A., & i Serra, R. S. (2022). Electroactive calcium-alginate/poly- caprolactone/reduced graphene oxide nanohybrid hydrogels for skeletal muscle tissue engineering. Colloids and Surfaces B: Biointerfaces, 214, 112455.
653. Chu, H., Johnson, N. R., Mason, N. S., & Wang, Y. (2011). A [polycation: heparin] complex releases growth factors with enhanced bioactivity. Journal of Controlled Release, 150(2), 157-163.
654. Awada, H. K., Johnson, N. R., & Wang, Y. (2014). Dual de- livery of vascular endothelial growth factor and hepatocyte growth factor coacervate displays strong angiogenic effects. Macromolecular bioscience, 14(5), 679-686.
655. Chu, H., Chen, C. W., Huard, J., & Wang, Y. (2013). The effect of a heparin-based coacervate of fibroblast growth factor-2 on scarring in the infarcted myocardium. Biomaterials, 34(6), 1747-1756.
656. Chu, H., Gao, J., Chen, C. W., Huard, J., & Wang, Y. (2011).Injectable fibroblast growth factor-2 coacervate for persis- tent angiogenesis. Proceedings of the National Academy of Sciences, 108(33), 13444-13449.
657. Lee, K. W., Johnson, N. R., Gao, J., & Wang, Y. (2013). Hu-
man progenitor cell recruitment via SDF-1α coacervate-laden PGS vascular grafts. Biomaterials, 34(38), 9877-9885.
658. Johnson, N. R., & Wang, Y. (2014). Coacervate delivery sys- tems for proteins and small molecule drugs. Expert opinion on drug delivery, 11(12), 1829-1832.
659. Black, K. A., Priftis, D., Perry, S. L., Yip, J., Byun, W. Y., & Tirrell, M. (2014). Protein encapsulation via polypeptide complex coacervation. ACS Macro Letters, 3(10), 1088-1091. 660.Oliveira, M. B., Ribeiro, M. P., Miguel, S. P., Neto, A. I., Coutinho, P., Correia, I. J., & Mano, J. F. (2014). In vivo high-content evaluation of three-dimensional scaffolds bio- compatibility. Tissue Engineering Part C: Methods, 20(11),
851-864.
661. Yu, J., Gu, Y., Du, K. T., Mihardja, S., Sievers, R. E., & Lee,R. J. (2009). The effect of injected RGD modified alginate
on angiogenesis and left ventricular function in a chronic rat infarct model. Biomaterials, 30(5), 751-756.
662. Rahman, M. S., Hasan, M. S., Nitai, A. S., Nam, S., Karmakar,A. K., Ahsan, M. S., ... & Ahmed, M. B. (2021). Recent devel- opments of carboxymethyl cellulose. Polymers, 13(8), 1345.
663. Silva, C. M., Ribeiro, A. J., Ferreira, D., & Veiga, F. (2006). Insulin encapsulation in reinforced alginate microspheres pre- pared by internal gelation. european journal of pharmaceuti- cal sciences, 29(2), 148-159.
664. Liu, M., Zeng, X., Ma, C., Yi, H., Ali, Z., Mou, X., ... & He,N. (2017). Injectable hydrogels for cartilage and bone tissue engineering. Bone research, 5(1), 1-20.
665. Bedian, L., Villalba-Rodríguez, A. M., Hernández-Vargas, G., Parra-Saldivar, R., & Iqbal, H. M. (2017). Bio-based mate- rials with novel characteristics for tissue engineering appli- cations–A review. International journal of biological macro- molecules, 98, 837-846.
666. Tang, Z. C., Liao, W. Y., Tang, A. C., Tsai, S. J., & Hsieh, P.C. (2011). The enhancement of endothelial cell therapy for angiogenesis in hindlimb ischemia using hyaluronan. Bioma- terials, 32(1), 75-86.
667. Grellier, M., Bordenave, L., & Amedee, J. (2009). Cell-to-cell communication between osteogenic and endothelial lineages: implications for tissue engineering. Trends in biotechnology, 27(10), 562-571.
668. Sun, G., Shen, Y. I., Ho, C. C., Kusuma, S., & Gerecht, S. (2010). Functional groups affect physical and biological prop- erties of dextranâ?based hydrogels. Journal of Biomedical Ma- terials Research Part A: An Oficial Journal of The Society for Biomaterials, The Japanese Society for Biomaterials, and The Australian Society for Biomaterials and the Korean Society for Biomaterials, 93(3), 1080-1090.
669. DeQuach, J. A., Lin, J. E., Cam, C., Hu, D., Salvatore, M. A., Sheikh, F., & Christman, K. L. (2012). Injectable skele- tal muscle matrix hydrogel promotes neovascularization and muscle cell infiltration in a hindlimb ischemia model. Europe- an cells & materials, 23, 400.
670. Rao, N., Agmon, G., Tierney, M. T., Ungerleider, J. L., Braden,R. L., Sacco, A., & Christman, K. L. (2017). Engineering an injectable muscle-specific microenvironment for improved cell delivery using a nanofibrous extracellular matrix hydro- gel. ACS nano, 11(4), 3851-3859.
671. Tanihara, M., Suzuki, Y., Yamamoto, E., Noguchi, A., & Miz- ushima, Y. (2001). Sustained release of basic fibroblast growth factor and angiogenesis in a novel covalently crosslinked gel of heparin and alginate. Journal of Biomedical Materials Re- search: An Oficial Journal of The Society for Biomaterials, The Japanese Society for Biomaterials, and The Australian Society for Biomaterials and the Korean Society for Biomate- rials, 56(2), 216-221.
672. Wissink, M. J. B., Beernink, R., Pieper, J. S., Poot, A. A., Engbers, G. H. M., Beugeling, T., ... & Feijen, J. (2001). Binding and release of basic fibroblast growth factor from heparinized collagen matrices. Biomaterials, 22(16), 2291-2299.
673. Lee, K. Y., Peters, M. C., Anderson, K. W., & Mooney, D. J. (2000). Controlled growth factor release from synthetic extra- cellular matrices. Nature, 408(6815), 998-1000.
674. Hong, Y., Chen, J., Fang, H., Li, G., Yan, S., Zhang, K., ... & Yin, J. (2020). All-in-One hydrogel realizing adipose-derived stem cell spheroid production and in vivo injection via “gel– sol” transition for angiogenesis in hind limb ischemia. ACS applied materials & interfaces, 12(10), 11375-11387.
675. Chen, X., Aledia, A. S., Ghajar, C. M., Griffith, C. K., Putnam,A. J., Hughes, C. C., & George, S. C. (2009). Prevasculariza- tion of a fibrin-based tissue construct accelerates the forma- tion of functional anastomosis with host vasculature. Tissue Engineering Part A, 15(6), 1363-1371.
676. Armulik, A., Genové, G., & Betsholtz, C. (2011). Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Developmental cell, 21(2), 193-215. 677.Song, H. H. G., Rumma, R. T., Ozaki, C. K., Edelman, E. R., & Chen, C. S. (2018). Vascular tissue engineering: progress, challenges, and clinical promise. Cell stem cell, 22(3), 340-
354.
678. Chandra, P., & Atala, A. (2019). Engineering blood vessels and vascularized tissues: technology trends and potential clin- ical applications. Clinical Science, 133(9), 1115-1135.
679. Chang, W. G., & Niklason, L. E. (2017). A short discourse on vascular tissue engineering. NPJ Regenerative medicine, 2(1), 7.
680. Lovett, M., Lee, K., Edwards, A., & Kaplan, D. L. (2009). Vascularization strategies for tissue engineering. Tissue Engi- neering Part B: Reviews, 15(3), 353-370.
681. Finking, G., & Hanke, H. (1997). Nikolaj Nikolajewitsch Anitschkow (1885–1964) established the cholesterol-fed rab- bit as a model for atherosclerosis research. Atherosclerosis, 135(1), 1-7.
682. Cimato, T., Beers, J., Ding, S., Ma, M., McCoy, J. P., Boehm, M., & Nabel, E. G. (2009). Neuropilin-1 identifies endothelial precursors in human and murine embryonic stem cells before CD34 expression. Circulation, 119(16), 2170-2178.
683. Hou, Q., Paul, A., & Shakesheff, K. M. (2004). Injectable scaf- folds for tissue regeneration. Journal of Materials Chemistry, 14(13), 1915-1923.
684. Mima, Y., Fukumoto, S., Koyama, H., Okada, M., Tanaka, S., Shoji, T., ... & Inaba, M. (2012). Enhancement of cell-based therapeutic angiogenesis using a novel type of injectable scaffolds of hydroxyapatite-polymer nanocomposite micro- spheres. PloS one, 7(4), e35199.
685. Sakiyama-Elbert, S. E., & Hubbell, J. A. (2000). Development of fibrin derivatives for controlled release of heparin-binding growth factors. Journal of controlled release, 65(3), 389-402. 686.Silva, E. A., Kim, E. S., Kong, H. J., & Mooney, D. J. (2008).
Material-based deployment enhances efficacy of endotheli- al progenitor cells. Proceedings of the National Academy of Sciences, 105(38), 14347-14352.
687. Nomura, Y., Sasaki, Y., Takagi, M., Narita, T., Aoyama, Y., & Akiyoshi, K. (2005). Thermoresponsive controlled asso- ciation of protein with a dynamic nanogel of hydrophobized polysaccharide and cyclodextrin: heat shock protein-like ac- tivity of artificial molecular chaperone. Biomacromolecules, 6(1), 447-452.
688. Miyata, T., Asami, N., & Uragami, T. (1999). A reversibly an- tigen-responsive hydrogel. Nature, 399(6738), 766-769.
689. Chen, Y., Wang, Z., & Zhou, L. (2014). Interleukin 8 Inhibi- tion Enhanced Cholesterol Efflux in Acetylated Low-Density Lipoprotein–Stimulated THP-1 Macrophages. Journal of In- vestigative Medicine, 62(3), 615-620.
690. Doran, A. C., Yurdagul Jr, A., & Tabas, I. (2020). Efferocyto- sis in health and disease. Nature Reviews Immunology, 20(4), 254-267.
691. Gerlach, B. D., Ampomah, P. B., Yurdagul, A., Liu, C., Lau- ring, M. C., Wang, X., ... & Tabas, I. (2021). Efferocytosis induces macrophage proliferation to help resolve tissue injury. Cell metabolism, 33(12), 2445-2463.
692. Singh, K. P., & Sharma, A. M. (2014). Critical limb ischemia: current approach and future directions. Journal of cardiovas- cular translational research, 7, 437-445.