

Research Article - (2022) Volume 6, Issue 2
The Collaborative Cross Mouse A Powerful Tool for Studying Complex Genetic Conditions of Neurogenesis and Brain Development
Received Date: Aug 02, 2022 / Accepted Date: Aug 08, 2022 / Published Date: Aug 10, 2022
Copyright: ©Copyright: Ã?©2022 Zhicheng Xiao. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Citation: Kuriakose, D., Xiao, Z. (2022). The Collaborative Cross Mouse â?? A Powerful Tool for Studying Complex Genetic Conditions of Neurogenesis and Brain Development. Stem Cell Res Int, 6(2), 54-63.
Abstract
Neurodegenerative disorders (NDD), like Alzheimer’s disease (AD), impose a large health and socio-economic burden. AD affected 50 million people worldwide in 2019, and was the fifth leading cause of death for people over 65 years old. Despite the high prevalences of NDDs, very little is known about their underlying mechanisms and causes. Commonly occurring comorbidities and symptoms – such as cognitive impairment – overlap, complicating differential diagnosis. Recent studies have also noted large differences between the diagnostic outcomes associated with different genetic causes. In humans, multiple genetic variants are associated with neurogenesis and NDDs. Most research on NDDs uses animal models that focus on a single mutation in a fixed genetic background, providing inadequate heterogeneity and power to map genes linked to complex disease conditions. Therefore, it is imperative for NDD research to use next-generation animal models with greater genetic diversity. The Collaborative Cross (CC) mouse captures over 90% of the genetic diversity of the mouse species in a set of recombinant inbred lines to maximize genetic power. The CC model improves the outcomes of lab research: it enables population-based laboratory study, generates efficient and reproducible datasets, and allows researchers to identify multiple causative genetic factors for neurobehavioural traits. The convergence of all these characteristics will advance the translational value of laboratory research and create commercially viable, cost-effective clinical interventions.
Keywords
Collaborative cross mice, Neurodegenerative disorders, Genetic diversity, Neurogenesis, Translational research, miR- NA, Animal models
Neurogenesis and Brain Development
Neurogenesis – the formation of new neurons in the brain – is cru- cial for the development of brain processes. Neurons differ great- ly in their structure and connections, which help them regulate functions in specific areas of the brain. Neurogenesis progresses through a set of neurogenetic events that occur during the early stages of brain development. The cells that mediate the neural fate are the neural progenitor cells (NPCs), which have stem cell-like properties. These stem cells give rise to new neurons, astrocytes and oligodendrocytes via neurogenesis and gliogenesis. Brain plasticity and neurogenesis are key to proper functioning of var- ious connections in embryonic and post-natal development. Any deviation from normal neurogenesis can contribute to psychiatric and NDDs.
Understanding the relationship between neuronal development and the hippocampus is critical to unravelling many NDDs, such as depression, Huntington’s disease and Alzheimer’s disease (AD). Neurogenesis in the hippocampus occurs primarily in the subventricular zone and dentate gyrus region of the brain. In the hippocampus, new neurons are formed from the neuroepithelium progenitor cells, which subsequently give rise to a granular cell layer. The neural stem cells present in the neurogenic niche differ- entiate to produce NPCs, after which they transform into neuro- blasts, and finally give rise to mature excitatory granule cells capa- ble of forming the neural circuitry required for proper functioning of the brain. The neural stem cells derived from both embryonic and adult brain regions have distinct genetic profiles due to the unique signals that activate them [1-3]. The function of neurogen- esis during embryonic and post-natal development is critical for processing of information related to different regions of the brain.
Adolescent Neurogenesis
Adolescence is an important stage for development of sound brain and body. It is a transition period into adulthood, considered criti- cal for development of proper adult behaviours and cognitive be- haviour. The adolescent period is considered to cover post-natal days 21–60 in mice and age group 12–18 years in humans [4]. During this stage there is increased capability for risk-taking, so- cial skills and impulsivity, which is attributed to the neurodevel- opmental changes occurring in the brain circuit machinery. The hippocampus is a hot site for these neuronal changes: increased numbers of granular cells and greater hippocampal volume have been observed during the adolescent stage. Moreover, hippocam- pal neurogenesis during the adolescent period is four times higher than within the dentate gyrus, and is evidenced by improvements in cognitive abilities like memory processing and pattern forma- tion [5, 6], preparing the brain for future challenges in adult life.
Altered lifestyle and physiological changes in this stage influence the formation of new neurons in the hippocampus. The adolescent body is subjected to a range of hormonal changes that can alter the pattern of neuronal proliferation and differentiation. Stress is detrimental to proliferation, causing differentiation of new neurons that reduce the overall number of neurons and cause cognitive dys- function. In addition, lifestyle factors like diet, gut microbiota (in- fluenced by diet) [7, 8], physical exercise and active learning are important regulators of neurogenesis. A distorted pattern of neuro- genesis can produce significant emotional instability, depression, mood disorders, schizophrenia or attention deficit hyperactivity disorder (ADHD) [9, 10]. All the factors outlined above determine the connectivity and functionality of hippocampal neurogenesis. Understanding neurogenesis during the adolescent period is there- fore critical for smooth transitioning to adulthood.
Adult Neurogenesis
The phenomenon of adult neurogenesis was first reported by Alt- man and Das (1965) in research on the hippocampus,[11] but was a matter of debate until recently, when Boldrini et al. [12] used markers to identify glial cells, NPCs, neuroblasts, and mature and immature neurons in the adult brain. The process of formation of new neurons involves a precursor stage, intermediate stage, matu- ration stage and survival stage [13]. The rate of neurogenesis dif- fers at various time points during the life course, and is attributed to changes in disease-modifying or environmental factors. Fur- thermore, evidence from animal studies shows functionally ac- tive neurons entering the brain circuitry. This ongoing neuronal development is believed to maintain cognitive function throughout adulthood and decline with impaired neurogenesis. Neurogenesis in the adult brain is also involved in functions like memory forma- tion, pattern segregation and mood regulation.
Neurodegenerative Disorders
Neurodegenerative disorders result from disrupted functioning of the neurological system, including of neurogenesis, synaptic con- nectivity and neuronal migration in the brain. Dysfunction of the brain structure can lead to psychiatric problems, learning impair- ments and emotional instability. NDDs may occur due to genetic, psychological and/or environmental factors. Genetic factors, like mutations in certain parts of genes that play a vital role in brain de- velopment, manifest as disease pathogenesis. A spectrum of muta- tions is associated with genes in the neurodevelopmental pathways related to transcriptional activity, chromatin remodelling, and syn- aptic function that lead to NDDs [14]. De novo mutations, copy number variations, single nucleotide polymorphisms have also been found to contribute to NDDs. Environmental factors like use of drugs, alcohol, exposure to chemicals and tobacco increase the risk of NDDs. Other non-genetic factors, like insults to endocrine and metabolic processes, can have profound effects on cognitive ability.
In adolescence, children can develop major mental illnesses such as ADHD, autistic spectrum disorder, learning and communication disorders, schizophrenia and bipolar disorder. NDDs that affect the older brain include AD, Parkinson’s disease, and amyotrophic lat- eral sclerosis. Furthermore, mutations in genes involved in devel- opment can alter cognitive ability in adulthood. This phenomenon can not only affect brain structure and function but produce cog- nitive phenotypes. Decreased neurogenesis is another important phenomenon that leads to NDDs. Alterations to neuronal pathways that supply energy for neurons and other cellular processes could lead to accumulation of peptide fragments within the brain that eventually reduce cognitive function [15].
Greater understanding of the dysfunctional pathways implicated in NDDs has paved the way for the development of many therapeutic drugs that target known pathways to minimise disease symptoms and restore function. They can recuperate the mutated gene or re- voke the signalling molecules in the affected areas of the brain, even after complete manifestation of the clinical symptoms [16]. However, it remains important to study the differences between adolescents and adults in the process of neurogenesis and the ge- netic machinery driving the formation of new neurons. Another treatment strategy intended to increase synaptic plasticity and im- mune modulation aims at boosting the metabolic capability of the cell to enhance formation of new brain cells and genetic machinery directed protein synthesis [15]. This could open a new horizon of therapeutic or diagnostic approaches that could enable better brain health during the entire life course.
Noncoding RNA
The transcriptome machinery of multicellular organisms is com- plex, and only recently has the role of ribonucleic acid (RNA) been studied extensively. More than two thirds of the genomic deoxyri- bonucleic acid (DNA) is transcribed, but less than 2% is translated into functional proteins. This means that the number of noncoding RNAs is higher than the number of protein genes within the cell. The noncoding RNA comprises microRNA (miR), short interfer- ing RNA (siRNA), PIWI-interacting RNA and long noncoding RNA. The length of noncoding RNA varies from >30 nucleotides (nt) for small and <200 nt for long noncoding RNA [17, 18].
MicroRNA
miRs are noncoding RNAs 18–25 nt long. miRs are usually lo- calised in the intronic or intergenic region of the host gene, and their expression levels correlate with that of the host gene [19, 20]. miR biogenesis starts with RNA polymerase II transcribing the long primary transcript (which is still part of mRNA) into pri- miR, which folds on itself to form the hairpin looped structure. This stem loop structure is cleaved by RNA III enzyme Drosha and DGCR8 to a 65 nt-long microprocessor complex called pre-miR (precursor miR). This process occurs in the nucleus, and the result is transported to the cytoplasm by the enzyme Exportin 5 [21, 22]. In the cytoplasm, pre-miR is broken down by the enzyme RNase III Dicer and transactivation response element RNAbinding pro- tein, resulting in a small RNA duplex. This complex is called the mature miR, which is then loaded into the RNA-induced silenc- ing complex with the help of Ago protein [23]. The complex is unwound to a single 18–25 nt long miR attributed to the helicase activity and chaperones within the cell [24-26]. This mature miR has a 6– 8 nt region, complementary to the target mRNA, which facilitates target gene degradation or translation inhibition within the cell. This entire process of miR biogenesis is Drosha enzyme dependent and hence is part of the canonical pathway. Another (Drosha-independent) pathway, called the non-canonical pathway RNA polymerase III, directly cleaves the pre-miR to mature miR, enabled by mirtrons [27].
miR-9
miR-9 is one of the most highly enriched miRs in the brain. It is conserved across the vertebrates, and is one of the most studied miRs, particularly due to its role in brain development and neu- rogenesis [28]. The presence of multiple copies of miR imparts to its vivid function within the cell [29]. Similar studies in other organisms like Xenopus, chick and zebrafish have revealed an im- portant role for miR-9 in the brain [29-32]. miR-9 drives the ex- pression of various mRNAs and transcription factors (TFs) to aid neurogenesis. The overexpression of miR-9 targets its downstream gene NR2E1 to promote renewal of neural stem cells and induce neural differentiation [33]. The expression of miR-9 in the cortex is important to regulate Foxp2 proteins, which hinder neuronal mi- gration and maturation of neurons in the cortex [34]. miR-9 also interacts with other miRs to maintain cell physiology and integrity. The increased level of miR107 in the cell decreases the expression of many miRs, of which miR-9 is the most affected [35]. miR-9 also interacts with miR-124 to produce functionally active neurons [36]. Huntington’s disease is linked to the transcriptional activity of restrictive element1 silencing transcription factor (REST) and coREST, which are targets of miR-9. The upregulation of these repressors is attributed to the decreased expression of miR-9 in diseased subjects compared to normal ones [37]. Similarly, miR-9 is downregulated in the brains of amyotrophic lateral sclerosis and AD patients, but upregulated in Parkinson’s patients [38-40].
miR-29a
miR-29a belongs to the miR-29 family, along with miR-29b and miR-29c. miR-29a is highly conserved in humans, rats and mice. The human brain is highly enriched in miR-29a, which plays an important role in neurogenesis and NDDs. BACE1 is a direct tar- get of miR-29a, and upon miR-29a downregulation the expression of the BACE1 gene is elevated, which contributes to plaque for- mation, as seen in AD patients [41]. miR-29a is upregulated in the hippocampus and cortex in the postnatal stages of development. There is strong evidence of increase in neural activity and miR- 29a expression in the primary neurons upon glutamate receptor activation. miR-29a is also expressed in the mature neurons. Dou- blecortin expression (DCX), a marker for neurons, is a direct target of miR-29a, and the transcriptional downregulation of DCX by miR-29a increases axonal branching [42, 43]. The miR-29a/b clus- ter targets ARCP3 protein to modulate synaptic plasticity; thus, the miR-29 family is important for neuronal differentiation, prolifera- tion and maturation within the cell. The upregulation of miR-29a decreases the expression of Vdac1 gene, which improves the apop- tosis and ataxia phenotype in brain cells [44].
miR-107
miR-107 belongs to the miR-103/miR-107 cluster and is highly expressed in the brain [45]. The miR-107 cluster regulates the ex- pression of Dicer, which controls the global expression of miRs within the cell. Differential regulation of miR-107 at the posttran- scriptional level affects neuronal cells. Upregulation of miR-107 interacts with the biogenesis of miR-430 and hinders normal brain morphology. Furthermore, downregulation of miR-107 causes increase in the expression of Dicer and promotes neurogenesis. Similarly, lower expression of miR-107 is attributed to increased activity of miR-9 [35]. miR-107 is a direct target of the BACE1 gene, and the expression of miR-107 decreases as AD progresses, hinting at a link between early disease pathology and miR-107 ex- pression level, suggesting this miR is a good candidate biomarker for AD diagnosis [46].
Transcription factors, miRs and mRNA loops
Transcription factors and miRs are vital in controlling gene ex- pression. They control transcription and post-transcriptional modification by targeting the mRNA to degrade or block protein synthesis. TFs bind to the promoter region of the target gene and control the rate of transcription [47]. The region on the mRNA, called the cis-regulatory DNA elements – where the TF binds – is the TF binding site, and is critical to process the information required to synthesise a functional protein. The network between TFmiR-mRNA forms a functional loop that is important for regu- lation of transcriptional and translational activities [48]. TF-miRs may be co-regulated to regulate their target mRNA or individually regulated upon external stimuli and modulate their downstream target [49, 50]. Many TFs have been identified so far, with spe- cific characteristics helping the cellular systems in various func- tions like signal transduction, posttranslational activities, ligand binding, and regulation of inflammatory responses [51]. They are specific to cell types and stages of development, and are critical in life cycle processes. The mode of action of TF depends upon the functional domains present. The effect can be positive or negative based on the interaction with the target gene. TFs can also interact with other TFs, co-activators, noncoding RNAs and chromatin re- modelling complexes.
The TFs can bind directly to the mRNA to regulate gene expres- sion. They bind to regulatory elements called the enhancers or si- lencers located in the 5′untranslated region of the target gene.
miRs are downstream targets of TFs. The regulation of gene ex- pression is exercised by binding to the 6–8 nt sequence located in the 5’ untranslated region, known as the seed region. Sometimes the seed region may not have an exact match to the first 6– 8 bases in the target site [52]. A single miR can target multiple mRNAs, and many miRs can bind to the target mRNA to initiate degrada- tion or destabilisation in their targets. miRs participate in various signalling activities and impose regulations against several excit- atory/inhibitory molecules that become susceptible to genetic or environmental perturbations. miRs are considered robust, hence miR biogenesis can be controlled by upstream signalling cues from TFs to modify target mRNA to provide crosstalk between signalling pathways. The TF–miR interaction occurs mainly in two steps: feed-forward loops (FFLs) and feedback loops (FBLs). FFL motifs co-regulate the TF–miR network by either TF regulat- ing miR or miR repressing the corresponding TF; the final effect will be a joint effort to degrade their target mRNA [48, 51]. FFLs can be further divided into two categories: coherent and incoherent FFLs. An FFL has two paths, one starting with TF as the master regulator, the second with miR as the master regulator. In the first case TF will regulate the expression of miR, and together they will drive the expression of their target; this is coherent FFL (Figure 1a). This kind of transcription is important to inhibit the leaky ex- pression of certain genes in a specific cell type. This mechanism can either activate or repress a particular gene [53]. Conversely, for incoherent FFL, the effect of the target gene is regulated by two opposite paths (Figure 1b). Here, miR is co-expressed with the tar- get gene, which helps it to check the expression level of the target, thereby reducing the effect of unwanted transcriptional signalling noise within the cell [54].
Feedback loops have a regulatory role in multiple organisms and occur mostly at the posttranscriptional level, as auto-regulatory loops [55-56]. In FBLs, TF-miRs regulate each other and inter- act with the target individually. They are single or double-negative FBLs based on how they interact with the target. In the single-neg- ative loop, a TF initiates the process of miR transcription, which in turn blocks the translation (Figure 1c). This process is important to control protein synthesis and maintain a proper cell cycle within the cell [57]. For the double-negative loop, TF-miRs act as toggle switches: when the TF is repressed, miR will be activated, and vice versa (Figure 1d). This type of regulation is vital to maintain long cellular responses and in cell differentiation [58, 59].
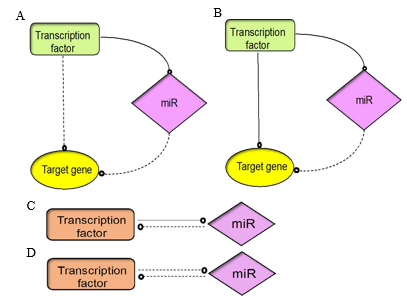
Figure 1: The architecture of TF and miR interaction depends on feedforward or feedback loops that control gene regulation. (A) Co- herent FFL, (B) Incoherent FFL (C) Single negative FBL, (D) Double negative FBL
Collaborative Cross Mice
The concept of Collaborative Cross (CC) or Gene mice emerged to overcome the restrictions of other recombinant inbred (RI) strains that lack adequate genetic heterogeneity and power to map genes linked to complex disease conditions. RI strains are typically derived from two close relatives and have little genetic diversity. However, humans differ greatly in their genetic archi- tecture, and a wide array of genetic, environmental and develop- mental factors contributes to complex disorders in humans, hin- dering the development of treatments. A well-developed animal model should provide the genetic diversity necessary to enable discovery of long-lasting solutions to complicated disorders like cancer, neurodegeneration, cardiovascular diseases, diabetes and obesity [60, 61]. Furthermore, it is difficult and expensive to de- velop a reproducible genetic model for characterisation and map- ping. Low availability of existing inbred strains and their ambig- uous heritability make them unreliable for genetic studies. Given all these problems, scientists decided to develop an animal model with maximum genetic diversity and reproducibility to maximise mapping power, and a reference population of CC mice was estab- lished [62].
Current Challenges
The science of human genetics has made considerable progress with the advent of techniques like genome-wide association stud- ies (GWAS). Many disease-specific loci have been identified, but in many cases the mechanisms directing these changes at the genome level are not elucidated clearly. The data obtained from GWAS is often not generalisable due to lack of genetic variation in questionable animal models. Due to the complex nature of the human body, many of the loci identified are casual genetic variants that fails to transition appropriately to clinical settings. In particu- lar, assaying the complex architecture of NDDs is overwhelming due to intertwined genetic network and disease progression. Fur- thermore, there is evidence of involvement of multiple factors in disease pathology. Therefore, the choice of a model organism to use to generate new knowledge about human disease is critical. Nonhuman counterparts can be used to build the knowledge re- quired to understand molecular pathways and genetic associations, but to draw scientific conclusions from animal studies, the genetic similarity between human and nonhuman equivalents should be strong. Single animal models often lack genetic variation com- pared to humans. This also affects the clinical translation of data derived from bench research. With advances in biological tech- niques like sequencing, computational biology and gene editing, and mouse genetics being studied comprehensively, animals can be developed and manipulated to generate a nonhuman population, with genetic diversity similar to that of the human population, for biological discovery.
CC Mice Strains
Collaborative Cross mice consist of a panel of RI mice lines derived from a genetically diverse set of eight founder strains comprising A/J, C57BL/6J, 129S1/SvlmJ, NOD/LtJ, NZO, CAST/Ei, PWK/ Ph, and WSB/Ei (as shown in Figure 2). These strains were select- ed from over 100 lines to capture maximum genetic diversity. The strains selected were those with genomes already sequenced (A/J, C57BL/6J, 129S1/SvlmJ), strains for disease models like diabetes, obesity, insulin resistance and immune dysregulation (NOD/LtJ, NZO) and wild-type strains (CAST/Ei, PWK/Ph, and WSB/Ei) to ensure highest genetic power [62, 63]. The breeding of CC mice gives access to an unlimited number of mice strains that can be manipulated in the laboratory to study different disease conditions and interventions. The reproducibility of these lines and the need to genotype the strains only once make this model even more at- tractive [60]. More than 1000 genetically diverse strains, all origi- nating from the eight founder strains, have been established.
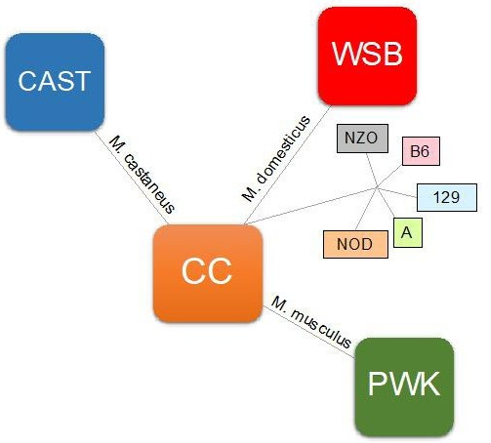
Figure 2: Phylogenetic tree of the eight CC founder strains. Red denotes M. domesticus origin; green, M. musculus origin; and blue, M. castaneus origin. Strains listed in dark coloured boxes are wild-type inbred strains.
Genetic Diversity of CC lines
The CC mouse was conceived as a platform for integrating re- producible phenotypic data and mapping mice population for complex traits. The genetic data from previous studies of mouse strains has hinted at the possibility of expansion of the phenotypic resources available in the laboratory mouse model [64]. For exam- ple, research conducted on Ebola virus susceptibility was possible because of the genetic diversity of CC mice [65]. Another feature of CC mice is the ability to track founder contributions using geno- typing platforms like MUGA and Mouse Diversity Array [66]. CC mice have also led to the advancement of various bioinformatics tools for haplotype reconstruction, genotyping arrays and devel- opment of new databases for storing of information related to the mouse phenotype. Sequencing analyses have indicated a change in the evolution of heterozygosity due to mutation and genetic drift in the CC lines over multiple periods of mating. The rate at which new mutations are observed is around 2.4 ± 0.4 per giga base per generation. Most of these mutations or polymorphisms are novel – observed only in the new generation of mice – and some could alter protein function at the transcriptome level [67].
The CC strains are capable of capturing approximately 135 re- combination events and polymorphisms every 100– 200 base pairs (bp) for high mapping resolution to generate statistical power to establish relevant biological interactions [60]. A two-stage map- ping process can increase efficacy and achieve higher mapping resolution. The first stage includes ~100 CC strains as a founda- tion panel to localise quantitative trait loci (QTL), whereas the sec- ond stage deploys additional information from another subset of ~100 CC lines to generate several recombination events and allelic patterns. The expected QTL mapping resolution is below 1 Mb. Genome-wide distribution studies on CC mice revealed three dis- tinct characteristics desirable in any laboratory mice population: balanced allele frequency, evenly distributed recombination sites, and genetic diversity [68]. The CC mouse also models the intri- cacies of the human genome and permits discovery of genotypic solutions to complex aetiologies originating from interactions be- tween environment and genetic conditions. It is the only mamma- lian database that supports high genome-wide variation across a large and heterogeneous population in a single laboratory and at a single point in time [60, 62, 68]. A typical CC mice line will resemble Figure 3 in terms of the genetic diversity it offers in any individual strain.

Figure 3: A typical CC line representing a genetic mosaic of the founder population derived from the crossbreeding of five inbred lines and three wildtype lines
Breeding Strategies
The eight founder CC mice strains were mated using funnel breed- ing to achieve a large number of RI lines with high genetic di- versity. A two-way, four-way and eightway crossing model was established for broader heritability of genetic data. The founder strains can be crossed in 56 unique ways to generate generation 1 (G1) crosses, in turn producing 1680 unique G2 crosses and final- ly 8! combinations for the G3 population. G3 mice carry genetic information from each of the founder strains and have maximum genetic power. After G3, 99% of the possible diversity will be achieved, and hence progress to the inbreeding stage with sibling (sib)-matings [62].
Breeding Stages
The three stages in the breeding process are mixing, maintenance and inbreeding (Figure 4). In the mixing stage, inbred strains are crossed to generate a foundation population of mice containing genetic information from each of the individual founder strains. In the maintenance stage, mice from the first stage are inter-crossed over a few generations to introduce numerous recombination events and produce mosaics of the founder mice population. It uses a pseudo-random mating technique to avoid mating two close- ly related mice, thereby reducing genetic drift. In the inbreeding stage, random mating occurs between two closely associated mice lines– sib-matings – for over 20 generations to generate discrete RI strains. The CC lines involve more of the mixing stage, following the funnel model of breeding. The mixing stage will produce AB, AC, … , FH, GH mice strains in a 2-way crossing. In the mainte- nance stage, AB × CD are crossed in a 4-way model, then breeding generates ABCDEFGH, called the foundation population of mice. Finally, the mice are inbred to fix the genetic variants [63].
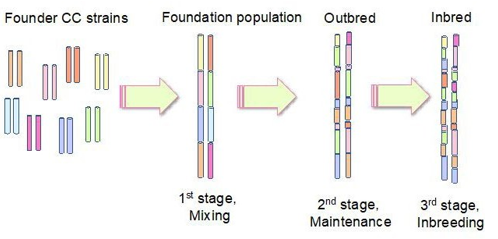
Figure 4: Breeding strategy of CC mice generating a mosaic cross. The first stage is mixing, which combines different inbred strains to produce a mouse population containing genetic material from each of the founder strains. In the maintenance stage, mice are inter-crossed to increase the number of recombination events. In the final inbreeding stage, recombinant inbred lines are generated.
CC Mice Research
Collaborative Cross lines served as a base model to produce a specialist genetically diverse mouse population, called Diversity Outbred, for high mapping resolution. It was seeded from the in- termediate generation of the CC mouse line and outbred through random matings to maximise genetic power [69, 70]. Furthermore, CC mice have enabled development of RI inter-crosses, which could stabilise phenotypic variations and provide a more fruitful model for understanding complex disorders [71]. Research using the CC mouse model will provide insight into the genes and iden- tification of new alleles at particular genomic locus in any complex disorders. CC mice have been used in research directed at targets as diverse as tumour susceptibility [72], human influenza [71], heart attack [73], neurodegeneration [74] and bone microarchitec- ture [75]. Thus, the CC mouse is a powerful tool for discovering novel pathways to genetic disorders.
CC Mice: a powerful resource for studying miR regulation and neurogenesis Hippocampal neurogenesis contributes to structural plasticity and neuronal integrity; conversely, a dysfunctional hip- pocampus results in memory impairment and cognitive disability [76]. The hippocampus is also vulnerable to hypoxia, stress and toxins, and is clinically relevant to NDDs resulting from hippo- campus-dependent behavioural deficits. Another unique feature of the hippocampus is lifelong adult neurogenesis. For all these reasons, dissecting the hippocampal signalling cues is critical for understanding AD. miRs of the miR-9/29a/107 family are expressed in the hippocam- pus and play a critical role in neurogenesis. Abrupt miR-9/29a/107 regulation may lead to AD pathology. Mechanisms of AD initi- ation and development are not well understood, and commonly occurring comorbidities and symptoms – such as cognitive impair- ment – overlap, complicating diagnosis. miRs, as biomarkers, offer great potential in AD research [43, 77-79]. A study have explored the upstream regulation of miR-9 and identified a transcriptional complex called miRSome through CC mice [80].
Recent studies have identified several miR-QTLs associated with complex disease phenotypes and underscore the importance of ge- netic regulation controlling miRs. GWAS have limited capability to extract information from any tissues in controlled environmen- tal conditions; CC mice can overcome these limitations [81]. Due to wellplanned breeding strategies, the genetic resolution of CC mice is substantial, providing more useful insights into molecular mechanism, disease conditions and various clinical traits than oth- er animal models. In addition, the underlying mechanisms linking differential gene expression, miR regulation and neurogenesis are poorly understood, while multiple factors and signalling pathways drive many processes additively or synergistically; these mecha- nisms and processes are hard to dissect using current laboratory animals due to restricted genetic diversity. CC mice provide gene expression data in a segregated population, helping to establish a relationship between complex genetic architecture and its environ- ment [82].
The general concept of animal models for NDDs focuses on a sin- gle mutation in a distinct genetic background. However, recent studies in human subjects have revealed that certain NDDs are not caused by single genetic mutations, but multiple variants. A further limitation to using a single animal model is that genotypic– pheno- typic relationships are not common to all genetic backgrounds. To overcome this hurdle, researchers adopted an approach that includ- ed the inbred mouse population, the outbred strains (descendants of inbred strains), and RI line panels to study genetic variation and furthermore to identify the QTLs associated with the disease phenotype. A disadvantage of this model is the need to genotype each mouse being studied, which can be expensive and time-con- suming. Another disadvantage is lower genetic diversity than the human population. The CC mouse reference population can give better resolution of the QTL for neurobehavioural traits. Research using CC mice to study NDDs has shown that this reference popu- lation can harbour phenotypic and genetic variability for multiple neurodevelopmental behaviors. It can also identify QTLs associat- ed with NDDs. Thus, the CC mouse is a powerful tool for studying genetic and environmental changes in a controlled genetic back- ground [74].
CC Mice and Translational Research
As many as 900,000 Australians may be suffering from dementia by 2050 [83]. AD accounts for 60–70% of dementia cases, making it one of the largest burdens on the health care system. The burden of AD extends beyond the cost of treating patients to the vast num- ber of work hours that must be diverted to carer responsibilities, and the psychological distress of family members as they watch their loved ones deteriorate. Finding effective treatments for these devastating diseases is of the utmost importance. The question is, is it possible to inhibit neurodegeneration by manipulating a sin- gle genetic factor or is it necessary to modulate multiple genetic factors for clinical intervention? Recent studies have revealed the polygenic nature of NDDs; that is, that multiple causative agents lead to NDDs.
The selection of animal models is critical for successful translation of research into clinical trials. As noted already, a single animal model does not adequately represent a complete disease condition occurring in the complex human brain. Animal models for NDDs mostly depend on a fixed genetic background, and the complexity observed in the genotype–phenotype relationship is not captured well. CC mice offer a diverse genetic background that allows us to identify multiple genetic factors contributing to a disease pheno- type, thus demonstrating the unique potential of population-based research.
Collaborative Cross mice move animal model studies beyond comparative single gene/single background designs to embrace multiple genetic backgrounds to quantify the effect of disease ae- tiologies. Utilising such multi-animal modelling in NDD research will be crucial for understanding the influence of genetic diversity on disease pathology and improving health outcomes.
Conclusion
For decades, mouse strains have provided an exquisite experimen- tal tool for studying the pathophysiology of disease and assessing therapeutic options in a genetically defined system. However, a major limitation of the mouse model is the low genetic diversity of common laboratory mice. CC mice represent a unique multi-an- imal model and a powerful innovative tool that allows capture of genetic diversity across mouse species, resembling a popula- tion-based study. The goal of any research is to translate scientific outcomes into clinical applications; the genetic diversity of CC mice greatly improves the practical potential of bench research. Notably, recent CC mice studies have identified multiple genet- ic factors contributing to NDD phenotypes [74]. CC mice can be used as a genetic reference population in complex gene expression studies, as well as research involving any tissue, any experimental or induced conditions, and CRISPR/Cas9-mediated genome edit- ing, to study the impact of genetic mutations across various genet- ic backgrounds.
Author Contributions: Conceptualization, D.K and Z.C.X; Writ- ing—Original Draft Preparation, D.K.; Writing—Review and Comments, Z.C.X.; Funding acquisition, Z.C.X.
Funding: Apex Biotech Research
Conflicts of Interest: The authors declare no conflict of interest.
References
- Hartenstein, V., & Stollewerk, A. (2015). The evolution of early neurogenesis. Developmental Cell, 32(4), 390-407.
- Kozareva, D. A., Cryan, J. F., & Nolan, Y. M. (2019). Born this way: hippocampal neurogenesis across the lifespan. Ag- ing Cell, 18(5), e13007.
- Berg, D. A., Su, Y., Jimenez-Cyrus, D., Patel, A., Huang, N., Morizet, D., ... & Bond, A. M. (2019). A common embryonic origin of stem cells drives developmental and adult neurogen- esis. Cell, 177(3), 654-668.
- Spear, L. P. (2000). The adolescent brain and age-related be- havioral manifestations. Neuroscience & biobehavioral re- views, 24(4), 417-463.
- Curlik, D. M., DiFeo, G., & Shors, T. J. (2014). Preparing for adulthood: thousands upon thousands of new cells are born in the hippocampus during puberty, and most survive with ef- fortful learning. Frontiers in neuroscience, 8, 70.
- Hueston, C.M., J.F. Cryan, and Y.M. Nolan, Stress and adoles- cent hippocampal neurogenesis: diet and exercise as cognitive modulators. Transl Psychiatry, 2017. 7(4): p. e1081.
- O’Mahony, S. M., Hyland, N. P., Dinan, T. G., & Cryan, J.F. (2011). Maternal separation as a model of brain–gut axis dysfunction. Psychopharmacology, 214(1), 71-88.
- Neufeld, K. A. M., Luczynski, P., Oriach, C. S., Dinan, T. G., & Cryan, J. F. (2016). What’s bugging your teen?—The microbiota and adolescent mental health. Neuroscience & Biobehavioral Reviews, 70, 300-312.
- Diaz-Granados, J. L., Greene, P. L., & Amsel, A. (1994). Se- lective activity enhancement and persistence in weanling rats after hippocampal X-irradiation in infancy: possible relevance for ADHD. Behavioral and neural biology, 61(3), 251-259.
- Reif, A., Schmitt, A., Fritzen, S., & Lesch, K. P. (2007). Neu- rogenesis and schizophrenia: dividing neurons in a divided mind?. European archives of psychiatry and clinical neurosci- ence, 257(5), 290-299.
- Altman, J., & Das, G. D. (1965). Autoradiographic and his- tological evidence of postnatal hippocampal neurogenesis in rats. Journal of Comparative Neurology, 124(3), 319-335.
- Boldrini, M., Fulmore, C. A., Tartt, A. N., Simeon, L. R., Pav-lova, I., Poposka, V., ... & Mann, J. J. (2018). Human hip- pocampal neurogenesis persists throughout aging. Cell stem cell, 22(4), 589-599.
- Kempermann, G. (2015). Activity dependency and aging in the regulation of adult neurogenesis. Cold Spring Harbor per- spectives in biology, 7(11), a018929.
- Niemi, M. E., Martin, H. C., Rice, D. L., Gallone, G., Gordon, S., Kelemen, M., ... & Barrett, J. C. (2018). Common genetic variants contribute to risk of rare severe neurodevelopmental disorders. Nature, 562(7726), 268-271.
- Gan, L., Cookson, M. R., Petrucelli, L., & La Spada, A. R. (2018). Converging pathways in neurodegeneration, from genetics to mechanisms. Nature neuroscience, 21(10), 1300- 1309.
- Ehninger, D., Li, W., Fox, K., Stryker, M. P., & Silva, A. J. (2008). Reversing neurodevelopmental disorders in adults. Neuron, 60(6), 950-960.
- Quinn, J. J., & Chang, H. Y. (2016). Unique features of long non-coding RNA biogenesis and function. Nature Reviews Genetics, 17(1), 47-62.
- Fatica, A., & Bozzoni, I. (2014). Long non-coding RNAs: new players in cell differentiation and development. Nature Reviews Genetics, 15(1), 7-21.
- Baskerville, S., & Bartel, D. P. (2005). Microarray profiling of microRNAs reveals frequent coexpression with neighboring miRNAs and host genes. Rna, 11(3), 241-247.
- Lau, N. C., Lim, L. P., Weinstein, E. G., & Bartel, D. P. (2001). An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science, 294(5543), 858-862.
- Bartel, D. P. (2009). MicroRNAs: target recognition and reg- ulatory functions. cell, 136(2), 215-233.
- Carthew, R. W., & Sontheimer, E. J. (2009). Origins and mechanisms of miRNAs and siRNAs. Cell, 136(4), 642-655.
- Chendrimada, T. P., Gregory, R. I., Kumaraswamy, E., Nor- man, J., Cooch, N., Nishikura, K., & Shiekhattar, R. (2005). TRBP recruits the Dicer complex to Ago2 for microRNA pro- cessing and gene silencing. Nature, 436(7051), 740-744.
- Iwasaki, S., Kobayashi, M., Yoda, M., Sakaguchi, Y., Katsu- ma, S., Suzuki, T., & Tomari, Y. (2010). Hsc70/Hsp90 chap- erone machinery mediates ATP-dependent RISC loading of small RNA duplexes. Molecular cell, 39(2), 292-299.
- Kim, V. N., Han, J., & Siomi, M. C. (2009). Biogenesis of small RNAs in animals. Nature reviews Molecular cell biolo- gy, 10(2), 126-139.
- Brodersen, P., & Voinnet, O. (2009). Revisiting the principles of microRNA target recognition and mode of action. Nature reviews Molecular cell biology, 10(2), 141-148.
- Berezikov, E., Chung, W. J., Willis, J., Cuppen, E., & Lai, E.C. (2007). Mammalian mirtron genes. Molecular cell, 28(2),328-336.
- Coolen, M., Katz, S., & Bally-Cuif, L. (2013). miR-9: a ver- satile regulator of neurogenesis. Frontiers in cellular neuro- science, 7, 220.
- Yuva-Aydemir, Y., Simkin, A., Gascon, E., & Gao, F. B.(2011). MicroRNA-9: functional evolution of a conservedsmall regulatory RNA. RNA biology, 8(4), 557-564.
- Darnell, D. K., Kaur, S., Stanislaw, S., Konieczka, J. K., Yatskievych, T. A., & Antin, P. B. (2006). MicroRNA expres- sion during chick embryo development. Developmental Dy- namics, 235(11), 3156-3165.
- Shibata, M., Kurokawa, D., Nakao, H., Ohmura, T., & Aiza- wa, S. (2008). MicroRNA-9 modulates Cajal–Retzius cell dif- ferentiation by suppressing Foxg1 expression in mouse me- dial pallium. Journal of Neuroscience, 28(41), 10415-10421.
- Bonev, B., Pisco, A., & Papalopulu, N. (2011). MicroRNA-9 reveals regional diversity of neural progenitors along the ante- rior-posterior axis. Developmental cell, 20(1), 19-32.
- Zhao, C., Sun, G., Li, S., & Shi, Y. (2009). A feedback regula- tory loop involving microRNA-9 and nuclear receptor TLX in neural stem cell fate determination. Nature structural & mo- lecular biology, 16(4), 365-371.
- Dajas-Bailador, F., Bonev, B., Garcez, P., Stanley, P., Guil- lemot, F., & Papalopulu, N. (2012). microRNA-9 regulates axon extension and branching by targeting Map1b in mouse cortical neurons. Nature neuroscience, 15(5), 697.
- Ristori, E., Lopez-Ramirez, M. A., Narayanan, A., Hill-Ter- an, G., Moro, A., Calvo, C. F., ... & Nicoli, S. (2015). A Di- cer-miR-107 interaction regulates biogenesis of specific miR- NAs crucial for neurogenesis. Developmental cell, 32(5), 546-560.
- Yoo, A. S., Sun, A. X., Li, L., Shcheglovitov, A., Portmann, T., Li, Y., ... & Crabtree, G. R. (2011). MicroRNA-mediated con- version of human fibroblasts to neurons. Nature, 476(7359), 228-231.
- Packer, A. N., Xing, Y., Harper, S. Q., Jones, L., & Davidson,B. L. (2008). The bifunctional microRNA miR-9/miR-9* reg- ulates REST and CoREST and is downregulated in Hunting- ton’s disease. Journal of Neuroscience, 28(53), 14341-14346.
- Cogswell, J. P., Ward, J., Taylor, I. A., Waters, M., Shi, Y., Cannon, B., ... & Richards, C. A. (2008). Identification of miRNA changes in Alzheimer’s disease brain and CSF yields putative biomarkers and insights into disease pathways. Jour- nal of Alzheimer’s disease, 14(1), 27-41.
- Kim, J., Inoue, K., Ishii, J., Vanti, W. B., Voronov, S. V., Mur- chison, E., ... & Abeliovich, A. (2007). A MicroRNA feedback circuit in midbrain dopamine neurons. Science, 317(5842), 1220-1224.
- Haramati, S., Chapnik, E., Sztainberg, Y., Eilam, R., Zwang, R., Gershoni, N., ... & Hornstein, E. (2010). miRNA malfunc- tion causes spinal motor neuron disease. Proceedings of the National Academy of Sciences, 107(29), 13111-13116.
- Roshan, R., Ghosh, T., Scaria, V., & Pillai, B. (2009). MicroR- NAs: novel therapeutic targets in neurodegenerative diseases. Drug discovery today, 14(23-24), 1123-1129.
- Bilimoria, P. M., De La Torre-Ubieta, L., Ikeuchi, Y., Beck- er, E. B., Reiner, O., & Bonni, A. (2010). A JIP3-regulated GSK3β/DCX signaling pathway restricts axon branching.Journal of Neuroscience, 30(50), 16766-16776.
- Li, H., Mao, S., Wang, H., Zen, K., Zhang, C., & Li, L. (2014).MicroRNA-29a modulates axon branching by targeting dou- blecortin in primary neurons. Protein & cell, 5(2), 160-169.
- Roshan, R., Shridhar, S., Sarangdhar, M. A., Banik, A., Chaw- la, M., Garg, M., ... & Pillai, B. (2014). Brain-specific knock- down of miR-29 results in neuronal cell death and ataxia in mice. Rna, 20(8), 1287-1297.
- Polster, B. J., Westaway, S. K., Nguyen, T. M., Yoon, M. Y., & Hayflick, S. J. (2010). Discordant expression of miR-103/7 and pantothenate kinase host genes in mouse. Molecular ge- netics and metabolism, 101(2-3), 292-295.
- Nelson, P. T., & Wang, W. X. (2010). MiR-107 is reduced in Alzheimer’s disease brain neocortex: validation study. Journal of Alzheimer’s Disease, 21(1), 75-79.
- Spitz, F., & Furlong, E. E. (2012). Transcription factors: from enhancer binding to developmental control. Nature reviews genetics, 13(9), 613-626.
- Shalgi, R., Lieber, D., Oren, M., & Pilpel, Y. (2007). Global and local architecture of the mammalian microRNA–tran- scription factor regulatory network. PLoS computational bi- ology, 3(7), e131.
- Somel, M., Guo, S., Fu, N., Yan, Z., Hu, H. Y., Xu, Y., ... &Khaitovich, P. (2010). MicroRNA, mRNA, and protein ex- pression link development and aging in human and macaque brain. Genome research, 20(9), 1207-1218.
- Li, A., Qin, G., Suzuki, A., Gajera, M., Iwata, J., Jia, P., & Zhao, Z. (2019). Network-based identification of critical reg- ulators as putative drivers of human cleft lip. BMC medical genomics, 12(1), 119-132.
- Zhang, H. M., Kuang, S., Xiong, X., Gao, T., Liu, C., & Guo,A. Y. (2015). Transcription factor and microRNA co-regulato- ry loops: important regulatory motifs in biological processes and diseases. Briefings in bioinformatics, 16(1), 45-58.
- Mullany, L. E., Herrick, J. S., Wolff, R. K., Stevens, J. R., Samowitz, W., & Slattery, M. L. (2018). MicroRNAâ?tran- scription factor interactions and their combined effect on tar- get gene expression in colon cancer cases. Genes, Chromo- somes and Cancer, 57(4), 192-202.
- Bartel, D. P., & Chen, C. Z. (2004). Micromanagers of gene expression: the potentially widespread influence of metazoan microRNAs. Nature reviews genetics, 5(5), 396-400.
- Osella, M., Bosia, C., Corá, D., & Caselle, M. (2011). The role of incoherent microRNA-mediated feedforward loops in noise buffering. PLoS computational biology, 7(3), e1001101.
- Thieffry, D., Huerta, A. M., Pérezâ?Rueda, E., & Colladoâ? Vides, J. (1998). From specific gene regulation to genomic networks: a global analysis of transcriptional regulation in Escherichia coli. Bioessays, 20(5), 433-440.
- Inui, M., Martello, G., & Piccolo, S. (2010). MicroRNA con- trol of signal transduction. Nature reviews Molecular cell bi- ology, 11(4), 252-263.
- O’Donnell, K. A., Wentzel, E. A., Zeller, K. I., Dang, C. V., &Mendell, J. T. (2005). c-Myc-regulated microRNAs modulateE2F1 expression. Nature, 435(7043), 839-843.
- Alon, U. (2007). Network motifs: theory and experimental ap- proaches. Nature Reviews Genetics, 8(6), 450-461.
- Lu, L., Zhou, L., Chen, E. Z., Sun, K., Jiang, P., Wang, L., ...& Wang, H. (2012). A novel YY1-miR-1 regulatory circuit in skeletal myogenesis revealed by genome-wide prediction of YY1-miRNA network. PloS one, 7(2), e27596.
- Churchill, G.A., et al. (2004). The Collaborative Cross, a community resource for the genetic analysis of complex traits. Nat Genet. 36(11): p. 1133-7.
- Williams, S. M., Haines, J. L., & Moore, J. H. (2004). The use of animal models in the study of complex disease: all else is never equal or why do so many human studies fail to replicate animal findings?. Bioessays, 26(2), 170-179.
- Morahan, G., Balmer, L., & Monley, D. (2008). Establish- ment of “The Gene Mine”: a resource for rapid identification of complex trait genes. Mammalian Genome, 19(6), 390-393.
- Valdar, W., Flint, J., & Mott, R. (2006). Simulating the col- laborative cross: power of quantitative trait loci detection and mapping resolution in large sets of recombinant inbred strains of mice. Genetics, 172(3), 1783-1797.
- Rogala, A. R., Morgan, A. P., Christensen, A. M., Gooch, T. J., Bell, T. A., Miller, D. R., ... & de Villena, F. P. M. (2014). The Collaborative Cross as a resource for modeling human disease: CC011/Unc, a new mouse model for spontaneous colitis. Mammalian genome, 25(3), 95-108.
- Rasmussen, A. L., Okumura, A., Ferris, M. T., Green, R., Feldmann, F., Kelly, S. M., ... & Katze, M. G. (2014). Host genetic diversity enables Ebola hemorrhagic fever pathogene- sis and resistance. Science, 346(6212), 987-991..
- Yang, H., Ding, Y., Hutchins, L. N., Szatkiewicz, J., Bell, T. A., Paigen, B. J., ... & Churchill, G. A. (2009). A customized and versatile high-density genotyping array for the mouse. Nature methods, 6(9), 663-666.
- Srivastava, A., Morgan, A. P., Najarian, M. L., Sarsani, V. K., Sigmon, J. S., Shorter, J. R., ... & de Villena, F. P. M. (2017). Genomes of the mouse collaborative cross. Genetics, 206(2), 537-556.
- Threadgill, D. W., Miller, D. R., Churchill, G. A., & de Vil- lena, F. P. M. (2011). The collaborative cross: a recombinant inbred mouse population for the systems genetic era. ILAR journal, 52(1), 24-31.
- Iancu, O. D., Darakjian, P., Walter, N. A., Malmanger, B., Oberbeck, D., Belknap, J., ... & Hitzemann, R. (2010). Ge- netic diversity and striatal gene networks: focus on the het- erogeneous stock-collaborative cross (HS-CC) mouse. BMC genomics, 11(1), 1-12.
- Threadgill, D. W., & Churchill, G. A. (2012). Ten years of the collaborative cross. G3: Genes| Genomes| Genetics, 2(2), 153-156.
- Rönnegård, L., & Valdar, W. (2011). Detecting major genetic loci controlling phenotypic variability in experimental cross- es. Genetics, 188(2), 435-447.
- Wang, P., Wang, Y., Langley, S. A., Zhou, Y. X., Jen, K. Y., Sun, Q., ... & Mao, J. H. (2019). Diverse tumour susceptibility in Collaborative Cross mice: identification of a new mouse model for human gastric tumourigenesis. Gut, 68(11), 1942- 1952.
- Salimova, E., Nowak, K. J., Estrada, A. C., Furtado, M. B.,McNamara, E., Nguyen, Q., ... & Rosenthal, N. A. (2019). Variable outcomes of human heart attack recapitulated in genetically diverse mice. NPJ Regenerative medicine, 4(1), 1-15.
- Molenhuis, R. T., Bruining, H., Brandt, M. J., Van Soldt, P.E., Abu-Toamih Atamni, H. J., Burbach, J. P. H., ... & Kas, M.J. (2018). Modeling the quantitative nature of neurodevelop- mental disorders using Collaborative Cross mice. Molecular autism, 9(1), 1-11.
- Levy, R., Mott, R. F., Iraqi, F. A., & Gabet, Y. (2015). Collab- orative cross mice in a genetic association study reveal new candidate genes for bone microarchitecture. BMC genomics, 16(1), 1-14.
- Disouky, A., & Lazarov, O. (2021). Adult hippocampal neuro- genesis in Alzheimer’s disease. Progress in molecular biology and translational science, 177, 137-156.
- Rajman, M., & Schratt, G. (2017). MicroRNAs in neural de- velopment: from master regulators to fine-tuners. Develop- ment, 144(13), 2310-2322.
- Xie, H., Zhao, Y., Zhou, Y., Liu, L., Liu, Y., Wang, D., ... & Yang, M. (2017). MiR-9 regulates the expression of BACE1 in dementia induced by chronic brain hypoperfusion in rats. Cellular Physiology and Biochemistry, 42(3), 1213-1226.
- Shu, B., Zhang, X., Du, G., Fu, Q., & Huang, L. (2018). Mi-croRNA-107 prevents amyloid-β-induced neurotoxicity and memory impairment in mice. International journal of molecu- lar medicine, 41(3), 1665-1672.
- Kuriakose, D., Iraqi, F., Morahan, G., & Xiao, Z. (2022). Iden- tification of a upstream transcription complex regulating miR- 9 expression during neurogenesis. 10.21203/rs.3.rs-1826909/ v1
- Nikpay, M., Beehler, K., Valsesia, A., Hager, J., Harper, M. E., Dent, R., & McPherson, R. (2019). Genome-wide identifi- cation of circulating-miRNA expression quantitative trait loci reveals the role of several miRNAs in the regulation of car- diometabolic phenotypes. Cardiovascular research, 115(11), 1629-1645.
- Que, E., James, K. L., Coffey, A. R., Smallwood, T. L., Al-bright, J., Huda, M. N., ... & Bennett, B. J. (2021). Genetic architecture modulates diet-induced hepatic mRNA and miR- NA expression profiles in Diversity Outbred mice. Genetics, 218(3), iyab068.
- Dementia in Australia 2021 report released. 2021.