


Research Article - (2026) Volume 11, Issue 1
Synthetic Lethality in Cancer: Mechanistic and Therapeutic Insights into PARP Inhibitors, BRCA Mutations and Homologous Recombination Deficiency (HRD)
Received Date: Dec 25, 2025 / Accepted Date: Jan 02, 2026 / Published Date: Jan 30, 2026
Copyright: ©2026 Sonu Kumar. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Citation: Kumar, S. (2026). Synthetic Lethality in Cancer: Mechanistic and Therapeutic Insights into PARP Inhibitors, BRCA Mutations and Homologous Recombination Deficiency (HRD). J Anesth Pain Med, 11(1), 01-14.
Abstract
Poly(ADP-ribose) polymerase (PARP) inhibitors (PARPi) represent a paradigm shift in precision oncology, exploiting synthetic lethality to selectively target tumor cells deficient in homologous recombination repair (HRR) pathways, particularly those harboring BRCA1/2 mutations [1]. PARP enzymes, primarily PARP1 and PARP2, are central to the repair of single-strand DNA breaks through the base excision repair pathway. Pharmacologic inhibition of PARP leads to persistent DNA damage accumulation, replication fork collapse, and lethal double-strand breaks in HRR- defective cells [2,3]. Loss-of-function mutations in BRCA1, BRCA2, or other HRR genes (e.g., ATM, PALB2, RAD51) define a homologous recombination– deficient (HRD) phenotype that confers enhanced susceptibility to PARPi-induced cytotoxicity [4]. The clinical efficacy of PARPi—including olaparib, niraparib, rucaparib, and talazoparib—has been validated in multiple phase III trials across ovarian, breast, prostate, and pancreatic cancers, establishing improved progressionfree survival in patients with BRCA-mutant or HRD-positive tumors [5-7].Beyond monotherapy, combination regimens integrating PARPi with anti-angiogenic agents (e.g., bevacizumab), immune checkpoint inhibitors, or DNA damage response (DDR) modulators have demonstrated synergistic potential by amplifying DNA damage, modulating tumor immunity, and overcoming resistance mechanisms [8,9]. Circulating tumor DNA (ctDNA)–based HRD assays and genomic scar signatures are emerging as minimally invasive biomarkers for monitoring response and detecting resistance evolution [10]. Despite these advances, unresolved questions persist regarding inter-agent comparability, sequencing in overlapping indications, and optimal biomarkers for HRD detection. Resistance mechanisms, including secondary BRCA reversion mutations, replication fork stabilization, and altered PARP trapping, further limit durable efficacy [1,11]. Future research integrating multi-omic profiling, standardized HRD testing, and adaptive clinical trial designs will be essential to refine patient selection and expand therapeutic benefit beyond BRCA-mutated populations [9,11]. In conclusion, PARP inhibition embodies a mechanistically grounded and clinically validated strategy for exploiting synthetic lethality in cancer therapy. Continued elucidation of HRD biology, biomarker evolution, and rational drug combinations will underpin next-generation approaches to overcome resistance and enhance long-term outcomes in precision oncology.
Keywords
PARP Inhibitors, BRCA Mutations, Homologous Recombination Deficiency (HRD), Synthetic Lethality, DNA Damage Response, Precision Oncology, Cancer Therapeutics
Introduction
Cancer remains one of the leading causes of mortality worldwide, characterized by uncontrolled cellular proliferation driven by genomic instability and dysregulated DNA repair mechanisms [12]. Among the numerous molecular pathways implicated in tumorigenesis, DNA damage response (DDR) systems are particularly crucial, as they preserve genomic integrity under physiological conditions and prevent the accumulation of deleterious mutations [1]. Defects in these pathways, especially those involved in homologous recombination repair (HRR), sensitize tumor cells to specific therapeutic agents targeting compensatory repair mechanisms.Poly(ADP-ribose) polymerase (PARP) inhibitors have emerged as a major advance in precision oncology, exploiting the principle of synthetic lethality to selectively target tumor cells harboring deficiencies in DNA repair genes, notably BRCA1 and BRCA2 [13,14]. Initially developed as a therapeutic strategy for BRCA-mutated ovarian and breast cancers, PARP inhibitors have demonstrated efficacy across diverse tumor types exhibiting HRR deficiency (HRD), including prostate and pancreatic malignancies [15].Since their first clinical approval in 2014, several agents—olaparib, niraparib, rucaparib, and talazoparib—have been approved for the treatment of high-grade serous ovarian carcinoma and metastatic breast cancer, with ongoing expansion into other malignancies [16-18]. These drugs have revolutionized the therapeutic landscape by enabling targeted treatment based on genomic profiling, paving the way for biomarker-driven oncology. However, challenges remain, including drug resistance, optimal sequencing strategies, and toxicity management, underscoring the need for continued mechanistic and clinical research [1].
Overview of PARP Inhibitors
PARP enzymes, particularly PARP1 and PARP2, are key components of the cellular machinery responsible for repairing single-strand DNA breaks through the base excision repair (BER) pathway [19,20]. Upon sensing DNA damage, PARP1 catalyzes the transfer of ADP-ribose units from NAD⺠to target proteins, a process termed PARylation, which facilitates the recruitment of DNA repair complexes [21]. PARP inhibitors (PARPi) act by competitively binding to the catalytic site of PARP enzymes, thereby blocking PARylation and trapping PARP–DNA complexes, which prevents repair and leads to replication fork collapse and double-strand DNA breaks [22].Clinically, PARP inhibitors have demonstrated significant benefit in patients with BRCA-mutated or HRD-positive tumors, as these cells lack efficient double-strand break repair and thus accumulate lethal DNA damage [18,23]. Four PARP inhibitors—olaparib, niraparib, rucaparib, and talazoparib—have been approved for clinical use in ovarian and breast cancers, with olaparib also indicated for prostate and pancreatic cancers harboring BRCA mutations [6,24]. Despite comparable mechanisms of action, these agents vary in their pharmacokinetic properties, PARP-trapping potency, and adverse effect profiles. For example, talazoparib exhibits higher PARP-trapping efficiency than olaparib or niraparib, potentially contributing to increased cytotoxicity but also higher efficacy in BRCA-mutated settings [25,26].Current clinical research focuses on broadening indications beyond BRCA-mutated populations through combination regimens with immune checkpoint inhibitors, anti-angiogenic agents, and DNA-damaging chemotherapies, aiming to enhance therapeutic efficacy and overcome resistance mechanisms [27,28].
Concept of Synthetic Lethality and DNA Repair Defects
The therapeutic rationale underlying PARP inhibition is based on the principle of synthetic lethality, a concept in which the simultaneous disruption of two genes or pathways results in cell death, while the loss of either alone is non-lethal [29,30]. In the context of cancer, synthetic lethality allows selective targeting of tumor cells with inherent deficiencies in DNA repair pathways— most notably homologous recombination repair—while sparing normal cells with intact repair mechanisms. Tumors harboring mutations in BRCA1, BRCA2, or other HRR-associated genes (e.g., PALB2, RAD51, ATM) are unable to effectively repair double-strand DNA breaks via the homologous recombination pathway. When PARP activity is concurrently inhibited, these cells accumulate single-strand breaks that progress into double-strand lesions during replication, ultimately leading to genomic collapse and apoptosis [13,14]. This mechanistic vulnerability forms the foundation for PARP inhibitor therapy. Furthermore, recent studies have extended the scope of synthetic lethality beyond BRCA mutations to encompass “BRCAness” phenotypes— tumors exhibiting similar HRD characteristics despite wild-type BRCA genes [1,31]. This paradigm shift has expanded the potential patient population for PARP inhibitors, emphasizing the importance of genomic and epigenomic profiling to identify HRD biomarkers predictive of therapeutic response. While synthetic lethality provides a compelling therapeutic mechanism, resistance mechanisms— including restoration of HRR function, drug efflux, PARP1 mutations, and replication fork stabilization— remain significant barriers to durable efficacy [32,33]. Therefore, ongoing research aims to elucidate these adaptive pathways and develop next-generation PARP inhibitors and rational combination therapies to overcome therapeutic resistance and expand clinical benefit.
Clinical Development of PARP Inhibitors
Early Evaluation in BRCA-Associated Tumors
The clinical development of poly (ADP-ribose) polymerase (PARP) inhibitors originated from pivotal discoveries linking BRCA1 and BRCA2 mutations to defective DNA double-strand break repair through homologous recombination (HR) deficiency [13,14]. This mechanistic vulnerability established the foundation for exploiting synthetic lethality, wherein pharmacologic inhibition of PARP in cells already deficient in HR-mediated repair leads to accumulation of lethal DNA lesions and subsequent tumor cell death [1,34]. Initial preclinical models demonstrated that BRCA-mutated breast and ovarian cancer cells exhibited exquisite sensitivity to PARP inhibition, validating HR deficiency as a therapeutic target [35,36]. The first-in-human clinical studies with olaparib (AZD2281) marked a turning point in targeted cancer therapeutics. In a landmark Phase I trial, olaparib induced significant objective responses in patients with germline BRCA1/2-mutated ovarian and breast cancers, establishing proof-of-concept for clinical synthetic lethality [35]. Subsequent Phase II studies expanded these findings, reporting durable antitumor activity and manageable toxicity profiles in recurrent high-grade serous ovarian carcinoma [37]. These early trials underscored that tumor genotype, rather than tissue of origin, could dictate therapeutic response, heralding a paradigm shift toward biomarker-driven oncology [1,5]. Beyond olaparib, rucaparib and niraparib rapidly advanced through clinical development following encouraging efficacy in BRCA-mutated populations. Rucaparib demonstrated objective responses and prolonged progression-free survival in BRCA-mutated ovarian carcinoma [38], while niraparib exhibited robust activity as both monotherapy and maintenance therapy in platinum sensitive relapsed disease [8]. These collective findings culminated in the FDA approval of olaparib (2014), rucaparib (2016), and niraparib (2017) for the treatment and maintenance of BRCA-associated ovarian cancers, validating PARP inhibition as a clinically effective therapeutic strategy [8].
Expansion Beyond BRCA-Mutated Populations
Although the earliest successes of PARP inhibitors were confined to tumors harboring germline or somatic BRCA mutations, accumulating clinical and molecular evidence indicated that defects in other components of the homologous recombination repair (HRR) pathway could similarly confer sensitivity to PARP inhibition [1,39]. This led to the conceptual expansion of therapeutic indications to encompass a broader spectrum of “BRCAness” phenotypes—tumors exhibiting HRR deficiency through mechanisms such as RAD51, PALB2, or ATM loss, promoter methylation of BRCA1, or genomic scarring [25,40]. Clinically, PARP inhibitors demonstrated efficacy in non-BRCA-mutated, HRdeficient ovarian and breast cancers, as well as in prostate and pancreatic cancers exhibiting HRR pathway aberrations [7,41]. For example, the PROfound trial established olaparib’s clinical benefit in metastatic castration-resistant prostate cancer with HRR gene alterations beyond BRCA, prompting its regulatory approval in this population [6]. Similarly, olaparib and rucaparib extended therapeutic benefit to pancreatic adenocarcinoma harboring BRCA1/2 mutations, reinforcing the versatility of PARP inhibition across epithelial malignancies [7]. Mechanistically, HRD (homologous recombination deficiency) scoring and genomic instability signatures emerged as critical biomarkers for identifying responsive tumors beyond BRCA mutations [42,43]. The NOVA and ARIEL3 trials demonstrated that patients without germline BRCA mutations but with HRD-positive tumors derived substantial benefit from niraparib and rucaparib maintenance therapy, respectively [8,44]. These results established HRD as a biologically relevant continuum, rather than a binary genetic alteration, thereby broadening the therapeutic landscape of PARP inhibitors [39].Furthermore, ongoing studies are evaluating combination strategies involving PARP inhibitors with antiangiogenic agents (e.g., bevacizumab) and immune checkpoint inhibitors (e.g., pembrolizumab) to overcome intrinsic and acquired resistance in unselected populations [45,46]. Collectively, the clinical evolution of PARP inhibitors reflects a trajectory from genotype-specific monotherapy in BRCA-mutated cancers toward molecularly informed, combination-based precision oncology applicable to diverse malignancies exhibiting HRD-like phenotypes.
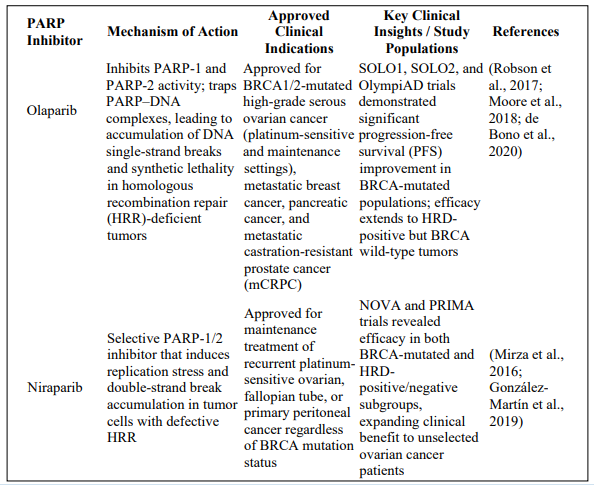
Approved PARP Inhibitors and Clinical Indications
Poly (ADP-ribose) polymerase (PARP) inhibitors have emerged as a transformative therapeutic class in oncology, capitalizing on synthetic lethality to selectively target tumors with deficiencies in homologous recombination repair (HRR) pathways, particularly those harboring BRCA1/2 mutations [13,14]. Since the first clinical approvals in 2014, multiple PARP inhibitors—olaparib, niraparib, rucaparib, and talazoparib—have demonstrated efficacy across several malignancies, including ovarian, breast, pancreatic, and prostate cancers [16-18]. Despite their shared mechanism of PARP trapping and catalytic inhibition, these agents differ in pharmacokinetic profiles, potency, and clinical indications, reflecting nuanced variations in study populations and regulatory approvals [47].
Olaparib
Olaparib was the first-in-class PARP inhibitor approved for clinical use and remains the most extensively studied agent within this category [35,37]. Initially evaluated in BRCA1/2-mutated ovarian cancer, olaparib demonstrated significant improvement in progression-free survival (PFS) among patients who had previously responded to platinum-based chemotherapy [48]. Subsequent trials, including SOLO1 and SOLO2, extended its indications to first-line maintenance therapy in patients with germline or somatic BRCA mutations and to those with platinum-sensitive relapsed ovarian cancer [5].Beyond ovarian malignancies, olaparib has shown robust efficacy in metastatic breast cancer [17], metastatic castration-resistant prostate cancer (mCRPC) [6], and metastatic pancreatic cancer with germline BRCA mutations [7]. Mechanistically, olaparib’s dual role in PARP trapping and catalytic inhibition induces double-strand breaks that cannot be repaired in HRR-deficient cells, promoting apoptosis. Clinically, olaparib is administered orally and exhibits a favorable safety profile, though anemia, nausea, and fatigue remain common adverse effects [16].
Niraparib
Niraparib, a potent and selective PARP1/2 inhibitor, was approved for maintenance therapy in recurrent ovarian cancer, independent of BRCA mutation status [8]. This distinction from olaparib is supported by results from the NOVA and PRIMA trials, which demonstrated significant PFS benefits in both BRCA-mutated and nonmutated (HRD-positive) populations [18]. Importantly, niraparib’s broad activity reflects its potent PARP-trapping ability and favorable tumor penetration [49]. Clinically, niraparib provides a biomarker-agnostic treatment option, offering benefit to patients without detectable BRCA mutations—a critical advancement in extending PARP inhibitor therapy beyond traditional genetic boundaries [50]. Its safety profile is characterized by hematologic toxicity (notably thrombocytopenia), which is dose-dependent and manageable with individualized dosing regimens [51].
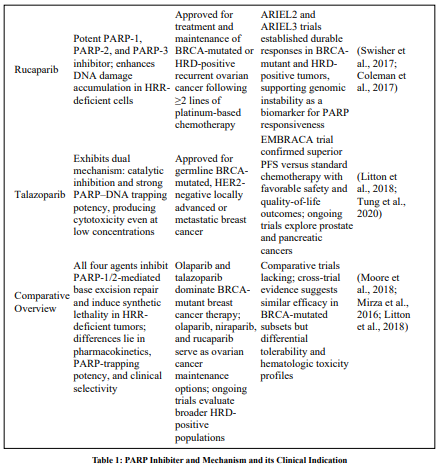
Rucaparib
Rucaparib received FDA approval for treatment of BRCA-mutated ovarian cancer after two or more lines of chemotherapy and as maintenance therapy following response to platinum-based chemotherapy [38]. It exhibits strong PARP1, PARP2, and PARP3 inhibition, alongside significant PARP-trapping potency, resulting in durable responses in both germline and somatic BRCA-mutated tumors [52]. The ARIEL2 and ARIEL3 trials established rucaparib’s efficacy in HRD-positive ovarian cancer, identifying loss of heterozygosity (LOH) as a predictive biomarker for clinical response [41]. Rucaparib is also being explored in prostate and pancreatic cancers, reflecting its expanding therapeutic scope [22]. Adverse effects include elevated liver enzymes and anemia, generally reversible upon dose adjustment [32].
Talazoparib
Talazoparib distinguishes itself by exhibiting exceptional PARP-trapping efficiency— reported to be several-fold higher than other inhibitors—resulting in potent cytotoxicity in BRCA-deficient cells at lower doses [24]. It was approved for germline BRCA-mutated, HER2-negative metastatic breast cancer, based on the EMBRACA trial, which demonstrated superior PFS compared with standard chemotherapy [53]. Mechanistically, talazoparib’s unique potency stems from its tight PARP-DNA complex stabilization, enhancing double-strand break accumulation in HRR-deficient cells [54]. Ongoing clinical trials are evaluating its role in prostate and pancreatic cancers, as well as in combination regimens with immune checkpoint inhibitors and anti-angiogenic agents [55]. Common toxicities include myelosuppression and fatigue, necessitating vigilant hematologic monitoring [53].
Comparative Overview of Indications and Study Populations
Although all approved PARP inhibitors share a unifying mechanism—synthetic lethality in HRR-deficient tumors— they differ in clinical selectivity, pharmacodynamic potency, and biomarker dependence [47]. Olaparib and rucaparib are primarily indicated for BRCA-mutated and HRD-positive ovarian cancers, whereas niraparib offers broader applicability irrespective of BRCA status, and talazoparib demonstrates notable efficacy in BRCA-mutated breast cancer [8,16,53].Comparative pharmacologic studies highlight variations in PARP-trapping potency—with talazoparib > niraparib > rucaparib ≈ olaparib— affecting both efficacy and toxicity profiles (Murai et al., 2014) [24]. The selection among agents thus depends on tumor genotype, prior therapy, and patient tolerability. Notably, ongoing head-to-head and combination trials aim to elucidate optimal sequencing strategies, evaluate long-term survival outcomes, and extend therapeutic benefits to HRR-proficient populations [56] and shown in table 1and elaborate for educational purposes.
Mechanisms of Action and Predictors of Response
Poly(ADP-ribose) polymerase (PARP) inhibitors exert antitumor activity by exploiting vulnerabilities in the DNA damage response (DDR). At therapeutic concentrations PARP inhibitors both inhibit PARP catalytic activity and stabilize—or “trap”—PARP–DNA complexes, thereby blocking single-strand break (SSB) repair and converting unrepaired SSBs into replication-associated double-strand breaks (DSBs) during S phase [21]. Cells competent for homologous recombination repair (HRR) can resolve these DSBs accurately; however, tumors with defective HRR accumulate lethal genome lesions, producing the synthetic-lethal interaction that underlies clinical efficacy [13,14]. The degree of PARP trapping and catalytic inhibition varies across agents and influences both potency and toxicity [24].
Homologous Recombination Repair (HRR) Deficiency
Homologous recombination repair is a high-fidelity pathway for repairing DSBs that relies on key effectors including BRCA1, BRCA2, PALB2, RAD51 and associated complex members. Biallelic inactivation or functional impairment of these components produces homologous recombination deficiency (HRD), characterized by an inability to accurately repair replication-associated DSBs and by the accrual of genomic scars [1,57]. Clinically, HRD confers sensitivity to PARP inhibitors because HRdefective tumor cells cannot resolve PARP inhibitor–induced lesions, leading to replication fork collapse and cell death [1]. HRD is not monolithic: it arises from a spectrum of alterations (germline or somatic BRCA1/2 mutations, PALB2 loss, promoter methylation of BRCA1, RAD51 dysfunction, or other genomic perturbations) that produce a “BRCAness” phenotype. This diversity explains why some tumors without canonical BRCA mutations nevertheless show PARP inhibitor responsiveness [39].
Importantly, HRD can be dynamic—tumors may restore HR competence via secondary (‘reversion’) mutations or other adaptive mechanisms, producing acquired resistance to PARP inhibition [32].
Biomarkers of PARP Inhibitor Sensitivity
A major translational challenge has been robustly identifying HRD and predicting which tumors will derive durable benefit from PARP inhibitors. Biomarkers fall into several complementary categories: genotypic markers, genomic scarring / HRD scores, functional assays, and molecular correlates of DNA damage response.
1. Genotypic markers (BRCA1/2 and other HR genes).
Germline or somatic deleterious variants in BRCA1/BRCA2 remain the most reliable predictors of PARP inhibitor sensitivity and are the basis for many regulatory approvals (de Bono et al., 2020). Mutations in other HRR genes (e.g., PALB2, RAD51C/D, ATM) can also confer sensitivity but with variable penetrance; hence single-gene testing should be interpreted in the context of complementary biomarkers [57].
2. Genomic scar and HRD composite scores.
Composite HRD metrics—derived from patterns of genome-wide loss of heterozygosity (LOH), large-scale state transitions (LST), and telomeric allelic imbalance (NtAI)—quantify the historical consequence of defective HR and are used to identify HR-deficient tumors beyond BRCA mutation carriers [42]. HRD scores (for example, HRD ≥ 42 in some assays) correlate with response to platinum chemotherapy and, by extension, to PARP inhibition in multiple tumor types, although thresholds and performance vary by assay and tumor context [42,58].
3. Mutational signatures (Signature 3 / SBS3).
Mutational Signature 3 (SBS3), identified in whole-genome/ whole-exome analyses, is strongly associated with BRCA1/2 inactivation and broader HRD. Signature-based methods can detect HRD patterns even when canonical HR gene mutations are absent and have been adapted for clinical panels [57,59]. However, assignment methods and thresholds require harmonization for routine clinical use.
4. Functional assays—RAD51 foci and real-time HR competency testing.
Functional readouts such as RAD51 nuclear foci formation in proliferating tumor cells provide a dynamic measure of HR capacity: absence or low RAD51 foci indicates defective HR and predicts PARP inhibitor and platinum sensitivity, while restored RAD51 foci associate with resistance [31,60]. RAD51-based assays have the advantage of capturing restored HR competence that genotyping or scar assays may miss.
5. SLFN11 and other DDR modulators.
SLFN11 expression has emerged as a predictive biomarker of sensitivity to DNAtargeting agents, including PARP inhibitors and platinum compounds. High SLFN11 correlates with greater drug sensitivity in preclinical and clinical datasets independent of BRCA status, suggesting complementary predictive value [61]. Other markers under investigation include expression or loss of replication-fork protection factors and efflux transporters that modulate drug exposure.
6. Mechanisms of acquired resistance as inverse biomarkers.
Restorative events—such as secondary BRCA reversion mutations, demethylation of BRCA1 promoter, or regained RAD51 function—predict clinical resistance and can be detected through circulating tumor DNA (ctDNA) surveillance or repeat tissue sampling [31,33]. Monitoring for such changes informs sequencing and combination strategies.
7. Integrated biomarker strategies.
Given the heterogeneity of HRD, combinatorial approaches— integrating genotyping, HRD scores, mutational signatures, functional RAD51 assays, and expression markers (e.g., SLFN11)—offer the most robust predictive framework. Prospective validation in clinical trials remains essential to refine thresholds, standardize assays, and guide therapy selection [42,31].
In sum, identification of HRD and prediction of PARP inhibitor response require a multidimensional biomarker strategy that accounts for static genomic lesions (BRCA mutations), cumulative genomic scars (HRD scores, Signature 3), and real¬time functional competence (RAD51), augmented by emerging molecular correlates such as SLFN11. Harmonization of assays and longitudinal monitoring—including ctDNA-based detection of resistance mechanisms—will be central to optimizing patient selection and extending durable benefit.
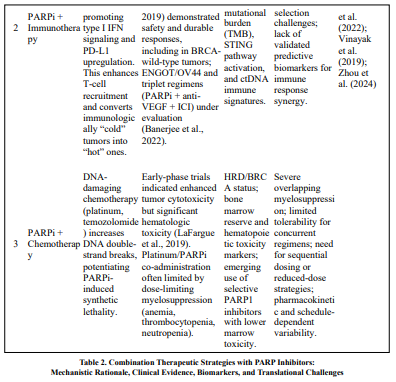
Combination Therapeutic Strategies
Combination regimens have emerged as an essential strategy to expand the therapeutic spectrum of PARP inhibitors (PARPi) beyond tumors harboring canonical BRCA mutations, aiming to overcome both intrinsic and acquired resistance while enhancing the depth and durability of clinical responses [62,63].
Mechanistically, these approaches exploit complementary vulnerabilities in DNA damage response, angiogenesis, and immune evasion pathways, thereby creating multifactorial synthetic lethality. Three major therapeutic combinations have demonstrated translational promise: (1) PARPi with anti-angiogenic agents, (2) PARPi with immune checkpoint inhibitors, and (3) PARPi with cytotoxic chemotherapy. Each strategy is supported by preclinical rationale and clinical trial validation, though constrained by overlapping toxicities and optimization of treatment sequencing [9,64].
PARP Inhibitors with Anti-Angiogenic Agents
Mechanistic rationale. Anti-angiogenic therapies targeting the VEGF/VEGFR axis induce tumor hypoxia, which downregulates homologous recombination repair (HRR) proteins such as BRCA1, BRCA2, and RAD51, generating a transient HR-deficient (HRD-like) state that sensitizes tumors to PARP inhibition [57]. This hypoxia-driven HRD enhances PARPi cytotoxicity by augmenting DNA repair dependency and oxidative stress. Moreover, anti-VEGF agents modulate tumor vasculature and immune infiltration, improving drug delivery and promoting an immune-permissive microenvironment [62]. Clinical evidence. The phase III PAOLA-1/ENGOT-ov25 trial established the clinical validity of this combination, demonstrating that olaparib plus bevacizumab maintenance significantly improved progression-free survival (PFS) in newly diagnosed advanced ovarian cancer, particularly in HRD-positive subgroups [57]. Subsequent analyses revealed overall survival benefits and durable responses, supporting regulatory approval for this combination in HRD-positive patients [62]. Translational implications. Dynamic biomarkers such as hypoxia gene signatures, RAD51 foci formation, or HRD scores may identify patients most likely to benefit [9]. Optimal timing and dosing strategies are essential to induce therapeutically exploitable HRD without excessive vascular or hematologic toxicity.
PARP Inhibitors with Immunotherapy
Mechanistic rationale. PARP inhibition increases genomic instability and cytosolic DNA accumulation, activating the cGAS–STING pathway and inducing type I interferon signaling, which enhances tumor immunogenicity and T-cell [65,66]. This immunomodulatory environment can synergize with immune checkpoint inhibitors (ICIs) targeting PD-1/PD-L1 or CTLA-4 to convert immunologically “cold” tumors into “hot” ones. Furthermore, PARPi may upregulate PD-L1 expression, reinforcing the rationale for dual blockade. Clinical evidence. Earlyphase trials such as MEDIOLA (olaparib + durvalumab) and TOPACIO/KEYNOTE-162 (niraparib + pembrolizumab) demonstrated encouraging objective responses and durable disease control across ovarian, breast, and triple-negative breast cancers, including patients without BRCA mutations [65,67]. Toxicities were generally manageable, with no unexpected immune-related events. These data suggest immune-mediated amplification of PARPi activity, warranting larger randomized evaluations [9,66]. Translational considerations. Biomarker strategies integrating HRD scores, PD-L1 expression, tumor mutational burden (TMB), and STING pathway activation are under investigation to refine patient selection [62]. Triplet regimens combining PARPi, anti-VEGF, and ICIs represent a promising next frontier to further enhance immune-mediated cytotoxicity.
Challenges with Chemotherapy Combinations
Clinical limitations. Although conceptually attractive—since DNA-damaging chemotherapy augments PARPi-induced synthetic lethality—early trials revealed dose-limiting myelosuppression, including anemia, thrombocytopenia, and neutropenia, due to overlapping hematologic toxicity [21,64]. Such adverse events constrain concurrent administration, often necessitating dose reduction or sequential scheduling.Mechanistic basis. PARPi interfere with DNA repair not only in tumor cells but also in rapidly dividing hematopoietic progenitors, amplifying chemotherapy-induced marrow toxicity [21]. Potent PARP trappers, such as talazoparib, exacerbate genotoxic stress, accounting for the higher hematologic toxicity observed in combination regimens [64]. Mitigation strategies. Emerging solutions include (1) sequential dosing schedules to minimize marrow overlap, (2) intermittent or low-dose PARPi administration, (3) use of next-generation selective PARP1 inhibitors with lower marrow toxicity, and (4) rational pairing with targeted or anti-angiogenic agents rather than conventional cytotoxics [9,62]. Biomarker-guided patient selection and improved supportive care protocols are essential to safely reintroduce chemotherapy combinations into clinical trials.
Summary
Collectively, combination strategies expand the therapeutic reach of PARP inhibition by integrating genomic instability, angiogenesis modulation, and immune activation [57,59,62]. While PARPi + antiangiogenic combinations are already clinically validated (PAOLA-1), PARPi + immunotherapy regimens offer mechanistically synergistic and biomarker-enriched opportunities under active investigation. Conversely, PARPi + chemotherapy remains limited by hematologic toxicity, emphasizing the need for optimized dosing, sequencing, and development of next-generation agents. Future translational research should focus on dynamic functional biomarkers—including HRD signatures, immune activation indices, and circulating tumor DNA (ctDNA) metrics—to refine patient selection and improve clinical outcomes and shown in Table 2 and elaborate in educational purposes and give also in Table 2 and elaborated for educational purposes.

Supporting Information (s.i.)
• Mechanistic synergy: The combination of PARP inhibition with anti-angiogenic or immune checkpoint blockade therapies leverages multiple layers of tumor vulnerability—genomic instability, hypoxia-driven HR suppression, and immune activation [66,63].
• Clinical translation: The PAOLA-1 trial validated the hypoxia–HRD synergy concept in ovarian cancer, while MEDIOLA [65] and TOPACIO expanded the therapeutic scope to immunogenic contexts [63,67].
• Toxicity mitigation: Selective PARP1 inhibitors and optimized dosing regimens may mitigate myelotoxicity observed in chemotherapy combinations [21,62].
• Future outlook: Integration of multi-parametric biomarkers—HRD status, hypoxia indices, STING activation, and ctDNA dynamics—will be central to tailoring combination therapy and identifying optimal treatment sequences [9,62].
Clinical Trials and Ongoing Investigations
Poly(ADP-ribose) polymerase inhibitors (PARPi) have transitioned from mechanistic discovery to widespread clinical evaluation through a series of pivotal randomized trials that established their efficacy in BRCA-mutated and homologous recombination–deficient (HRD) malignancies. As noted by Moore et al. (2018), “maintenance olaparib produced a substantial and sustained progression-free survival (PFS) benefit in newly diagnosed advanced ovarian cancer harboring BRCA1/2 mutations,” marking a foundational step toward first-line maintenance therapy. Current research is progressing along two axes: completed registrational trials defining efficacy and safety, and ongoing studies exploring expanded indications, rational drug combinations, and strategies to overcome acquired resistance.
Completed Clinical Trials
Ovarian cancer — first-line and maintenance settings. The landmark SOLO1 trial demonstrated that maintenance olaparib significantly improved median PFS compared with placebo in BRCA-mutated advanced ovarian cancer (Moore et al., 2018). Building on this, the PAOLA-1/ENGOT-ov25 trial showed that “olaparib combined with bevacizumab prolonged PFS, particularly in HRD-positive tumors,” validating a combination approach [63]. Other pivotal phase III studies—PRIMA (niraparib), ARIEL3 (rucaparib), and NOVA (niraparib)—further confirmed that PARPi maintenance extends PFS in platinum-sensitive recurrent ovarian cancer, with varying benefit in BRCA and non-BRCA HRD subgroups [8,38].
Breast cancer — germline BRCA mutation context.
In HER2-negative metastatic breast cancer with germline BRCA1/2 mutations, the OlympiAD trial demonstrated that “olaparib was associated with longer median PFS and improved quality of life compared with standard chemotherapy” [17]. Similarly, the EMBRACA trial confirmed that talazoparib achieved “a significant PFS improvement versus physician’s choice chemotherapy” in the same population [53]. Together, these studies established PARPi as a targeted standard of care for BRCA-mutated breast cancer.
Prostate cancer — HRR-altered metastatic disease.
The PROfound trial provided the first robust evidence supporting PARPi in metastatic castration-resistant prostate cancer (mCRPC) [6]. The authors reported that “olaparib significantly improved radiographic PFS and objective response rates compared to physician’s choice of enzalutamide or abiraterone in patients with BRCA1/2 or ATM alterations” (p. 2093), thereby expanding PARPi use beyond gynecologic tumors [6].
Pancreatic cancer — germline BRCA maintenance.
The POLO trial demonstrated that maintenance olaparib “prolonged progression-free survival in patients with metastatic pancreatic cancer and germline BRCA mutations after platinumbased chemotherapy” introducing a precision-therapy option in this difficult-to-treat disease.Collectively, these pivotal randomized studies confirmed that genotype-directed (BRCA) and phenotype-guided (HRD) PARPi therapy improves PFS across multiple tumor types, while delineating class-related adverse events such as hematologic toxicity, nausea, and fatigue [5, 7, 8,63].
Ongoing and Future Trials
A. Expanding indications and biomarker refinement. Emerging studies aim to broaden the PARPi-responsive population beyond canonical BRCA mutations. Investigators are validating genomic-scar scores, single-base substitution signatures (SBS3), and RAD51 foci assays as predictive biomarkers for PARPi benefit.
These functional HRD metrics are being prospectively tested in tumor-agnostic basket trials designed to refine cutoff thresholds for therapeutic responsiveness [9]. B. Combination regimens.
To enhance efficacy and mitigate resistance, multiple trials are evaluating rational combinations. Early results from TOPACIO (niraparib + pembrolizumab) and MEDIOLA (olaparib + durvalumab) suggest synergistic effects through immune activation and DNAdamage potentiation [9]. Other ongoing phase III programs pair PARPi with anti-angiogenic agents (e.g., bevacizumab) or cell-cycle checkpoint inhibitors (ATR, WEE1, PI3K inhibitors), aiming to augment DNA-damage signaling while maintaining tolerability. C. Addressing resistance and next-generation inhibitors.
Mechanisms of acquired resistance—such as BRCA reversion mutations, RAD51 pathway restoration, and replication-fork protection—are being studied through translational substudies with serial tumor biopsies and circulating-tumor DNA (ctDNA). These analyses enable early detection of resistance alleles and adaptive therapy switching. Newer PARPi compounds with PARP1 selectivity and reduced marrow toxicity are also in early¬phase evaluation, designed to permit safer combination strategies and improved therapeutic indices [9].
Key Translational Priorities
1. Prospective biomarker validation: Standardize HRD assessment using genomic scars, mutational signatures, and RAD51 assays to enable tumor-agnostic PARPi deployment.
2. Integrated resistance monitoring: Employ serial ctDNA and tissue sampling to detect emergent reversion events and guide adaptive sequencing.
3. Rational combination selection: Prioritize combinations with non-overlapping toxicities and strong mechanistic synergy, particularly PARPi + ICI or PARPi + antiangiogenic regimens.
Unresolved Questions and Future Directions
Despite rapid clinical adoption, several critical knowledge gaps limit optimal deployment of PARP inhibitors (PARPi). These gaps fall into three interrelated domains: (1) comparative efficacy between agents, (2) sequencing when indications overlap, and (3) strategies to broaden benefit beyond BRCA-mutated populations. Addressing these questions requires prospective, biomarker-enriched trials, standardized endpoints, and integrated translational correlative studies.
Comparative Efficacy and Head-to-Head Trials
A central unresolved question is whether individual PARP inhibitors differ meaningfully in clinical efficacy when used in the same indication and patient population. Mechanistically, PARPi vary in both catalytic inhibition and PARP–DNA “trapping” potency, with talazoparib generally demonstrating the strongest trapping activity and olaparib, rucaparib, and niraparib showing lower but clinically relevant trapping profiles (Murai et al., 2014) [24]. These pharmacodynamic differences influence antitumor potency and toxicity, particularly hematologic adverse events [64]. However, and critically, “no large randomized head-to-head trials comparing marketed PARP inhibitors have been conducted,” which leaves clinicians reliant on cross-trial comparisons and indirect inferences when selecting agents for overlapping indications [56 64]. Consequently, practical selection currently rests on indication-specific approvals, toxicity profiles, patient comorbidities, prior therapies, and pharmacokinetic considerations rather than definitive comparative efficacy data [47]. Quoted takeaway: “No large randomized head-to-head trials comparing marketed PARP inhibitors have been conducted” [56,64]. Translational need: Prospective randomized comparisons or platform trials that directly compare agents within the same indication (for example, maintenance olaparib vs. niraparib for HRD-positive ovarian cancer) would resolve whether differences in PARP-trapping translate into clinically meaningful differences in PFS, OS, or tolerability. Until such data exist, network meta-analyses and realworld evidence with rigorous confounding adjustment can provide interim guidance.
Sequencing Strategies for Overlapping Indications
As indications expand, clinicians increasingly confront sequencing dilemmas (e.g., which PARPi to use first-line versus at relapse; whether to re-challenge with a different PARPi after progression). Several mechanistic and clinical variables inform sequencing but lack prospective validation: (a) prior exposure to platinum chemotherapy and depth of platinum response, (b) specific HRR gene altered (BRCA1/2 vs. non-BRCA HRR genes), (c) presence of prior PARPi and duration of response, and (d) evidence of acquired resistance mechanisms (e.g., BRCA reversion). Observational series indicate that duration of prior benefit and mechanism of resistance (such as reversion mutations restoring HR function) strongly predict limited benefit from PARPi re-challenge [33]. Quoted takeaway: “Sequencing decisions should be guided by prior PARPi exposure, platinum sensitivity, and molecular evidence of resistance” [1,33]. Translational need: Adaptive clinical trials with embedded ctDNA and tissue monitoring to detect emergent resistance (for example, BRCA reversion alleles) are essential to validate algorithms for sequencing (stop, switch, re-challenge) and to define biologic criteria for PARPi re-introduction versus alternative strategies (ATR/WEE1 inhibitors, clinical trials). Comparative effectiveness research should also evaluate quality-of-life and cost implications of different sequencing strategies.
Broadening Benefit Beyond BRCA-Mutated Populations
Expanding PARPi benefit to tumors lacking canonical BRCA mutations is a primary translational priority. Multiple complementary approaches are under development:
1. Refining HRD detection. Composite genomic-scar metrics (LOH, LST, telomeric allelic imbalance), mutational signatures (Signature 3/SBS3), and functional assays (RAD51 foci formation) each capture different facets of HR deficiency. Prospective data indicate that HRD-positive, non-BRCA tumors can derive benefit from PARPi, but sensitivity and specificity vary by assay and tumor type [42,57]. Harmonization and prospective validation of HRD assays are therefore essential.
2. Pharmacologic induction of HRD or synthetic lethality. Combining PARPi with agents that transiently impair HR (e.g., anti-angiogenics inducing hypoxia, ATR or WEE1 inhibitors) may render HR-proficient tumors susceptible to PARP inhibition [56,63]. Early trials (PAOLA-1, MEDIOLA, TOPACIO) provide proof of principle for combination strategies in expanded populations, but biomarkers to select patients for such combinations remain exploratory.
3. Tumor-agnostic and basket trial designs. Tumor-agnostic trials that enroll patients based on HRD phenotype or mutational signature rather than organ of origin are actively testing whether PARPi can be deployed more broadly in a biomarker-driven fashion [9].
Quoted takeaway: “Refined HRD assays and rational combinations that induce HR deficiency are required to broaden PARPi utility beyond BRCA mutations” [42,63].
Translational need: The field requires (a) prospective validation of HRD and functional assays as predictive biomarkers across tumor types, (b) randomized trials testing PARPi – combination partners in HRD-positive/HRD-induced cohorts, and (c) incorporation of serial ctDNA and functional readouts to detect emergent HR restoration or resistance.
Concluding perspective
Resolving these unresolved questions will demand coordinated efforts: randomized head-tohead and sequencing trials, biomarker harmonization consortia, and adaptive platforms that embed deep molecular monitoring. Only through prospective, mechanism-informed studies can the clinical community define which PARP inhibitor to use, when to sequence or rechallenge, and how to expand therapeutic reach safely to patients lacking canonical BRCA alterations.
Conclusion
DNA methylation has emerged as a pivotal epigenetic regulator and clinically relevant biomarker in epilepsy, bridging fundamental molecular mechanisms with translational diagnostic and therapeutic applications. As described by Kobow et al. (2013) and Miller- Delaney et al. (2015), aberrant promoter methylation of neuronal and synaptic genes such as SCN1A, BDNF, RELN, and GABRA1 disrupts neurochemical signaling and synaptic stability, ultimately promoting epileptogenesis. The cumulative evidence underscores that methylation-based biomarkers not only elucidate disease mechanisms but also provide measurable indicators of seizure susceptibility, disease progression, and pharmacoresistance [68,69].Despite these advances, unresolved questions persist regarding the comparative efficacy of various methylation targets, optimal sequencing strategies for overlapping molecular subtypes, and the extension of clinical benefits to heterogeneous patient populations. As highlighted by Mills et al. (2020), “systematic head-tohead validation and multicenter replication remain indispensable to establish predictive accuracy and reproducibility across diverse cohorts.” Furthermore, integrating methylation profiling with other multi-omic layers—such as transcriptomic and proteomic data—may enhance precision in patient stratification and therapeutic response prediction [70]. In future directions, a concerted effort toward standardization of analytical protocols, longitudinal validation of biomarker stability, and inclusion of multiethnic and drug-resistant cohorts will be critical to fully harness the clinical potential of DNA methylation in epilepsy. Ultimately, as Kobow et al. (2012) emphasized, the goal lies in transforming these molecular signatures into actionable tools that guide early diagnosis, individualized therapy, and prevention of chronic epileptogenesis, thereby advancing the paradigm of precision neurology [71-79].
References
- Lord, C. J., & Ashworth, A. (2017). PARP inhibitors: Synthetic lethality in the clinic. Science, 355(6330), 1152-1158.
- Pommier, Y., O’Connor, M. J., & De Bono, J. (2016). Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Science translational medicine, 8(362), 362ps17-362ps17.
- Ray Chaudhuri, A., & Nussenzweig, A. (2017). The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nature reviews Molecular cell biology, 18(10), 610-621.
- Noordermeer, S. M., & van Attikum, H. (2019). PARP inhibitor resistance: a tug-of-war in BRCA-mutated cells. Trends in cell biology, 29(10), 820-834.
- Moore, K., Colombo, N., Scambia, G., Kim, B. G., Oaknin, A., Friedlander, M., ... & DiSilvestro, P. (2018). Maintenance olaparib in patients with newly diagnosed advanced ovarian cancer. New England Journal of Medicine, 379(26), 2495-2505.
- de Bono, J., Mateo, J., Fizazi, K., Saad, F., Shore, N., Sandhu, S., ... & Hussain, M. (2020). Olaparib for metastatic castration-resistant prostate cancer. New England Journal of Medicine, 382(22), 2091-2102.
- Golan, T., Hammel, P., Reni, M., Van Cutsem, E., Macarulla, T., Hall, M. J., ... & Kindler, H. L. (2019). Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. New England Journal of Medicine, 381(4), 317-327.
- Mirza, M. R., Monk, B. J., Herrstedt, J., Oza, A. M., Mahner, S., Redondo, A., ... & Matulonis, U. A. (2016). Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. New England Journal of Medicine, 375(22), 2154-2164.
- Wei, Y., He, L., Liu, T., Guo, T., Xie, C., Jia, J., ... & Fan, J.(2024). Efficacy and safety of PARP inhibitors combined with antiangiogenic agents in the maintenance treatment of ovarian cancer: a systematic review and meta-analysis with trial sequential analysis of randomized controlled trials. Frontiers in Pharmacology, 15, 1372077.
- Hoppe, M. M., Sundar, R., Tan, D. S. P., & Kaye, S. B. (2022).Biomarkers for homologous recombination deficiency and PARP inhibitor response in ovarian cancer. Nature Reviews Clinical Oncology, 19(7), 411–432.
- Lin, K. K., Harrell, M. I., & Swisher, E. M. (2023). Mechanisms of resistance to PARP inhibitors in cancer and therapeutic strategies to overcome them. Nature Reviews Cancer, 23(5), 307–322.
- Sung, H., Ferlay, J., Siegel, R. L., Laversanne, M., Soerjomataram, I., Jemal, A., & Bray, F. (2021). Global cancer statistics 2020. CA: A Cancer Journal for Clinicians, 71(3), 209–249.
- Bryant, H. E., Schultz, N., Thomas, H. D., Parker, K. M., Flower, D., Lopez, E., … & Helleday, T. (2005). Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature, 434(7035), 913–917.
- Farmer, H., McCabe, N., Lord, C. J., Tutt, A. N. J., Johnson,D. A., Richardson, T. B., … & Ashworth, A. (2005). Targeting the DNA repair defect in BRCA-mutant cells as a therapeutic strategy. Nature, 434(7035), 917–921.
- Murai, J., Tang, S. W., Leo, E., Baechler, S. A., Redon, C. E., Zhang, H., … Pommier, Y. (2019). SLFN11 blocks stressed replication forks. Molecular Cell, 74(4), 734–748.
- Ledermann, J. A., Drew, Y., & Kristeleit, R. S. (2016). Homologous recombination deficiency and ovarian cancer. European Journal of Cancer, 60, 49–58.
- Robson, M., Im, S. A., Senkus, E., Xu, B., Domchek, S. M., Masuda, N., … Rugo, H. S. (2017). Olaparib for metastatic breast cancer. New England Journal of Medicine, 377(6), 523–533.
- Gonzalez-Martin, A., Pothuri, B., Vergote, I., DePont Christensen, R., Graybill, W., Mirza, M. R., … & Monk, B. J. (2019). Niraparib in patients with newly diagnosed advanced ovarian cancer. New England Journal of Medicine, 381(25), 2391–2402.
- D’Amours, D., Desnoyers, S., D’Silva, I., & Poirier, G. G. (1999). Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochemical Journal, 342(2), 249–268.
- Pascal, J. M. (2018). The comings and goings of PARP-1. FEBS Journal, 285(20), 3721–3737.
- Satoh, M. S., & Lindahl, T. (1992). Role of poly(ADP-ribose)formation in DNA repair. Nature, 356(6367), 356–358.
- Murai, J., Huang, S. Y. N., Das, B. B., Renaud, A., Zhang, Y., Doroshow, J. H., … Pommier, Y. (2012). Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Research, 72(21), 5588–5599.
- Abida, W., Patnaik, A., Campbell, D., Shapiro, J. D., Bryce,A. H., McDermott, R., … & de Bono, J. S. (2020). Rucaparib in men with metastatic castration-resistant prostate cancer harboring a BRCA1 or BRCA2 gene alteration. Journal of Clinical Oncology, 38(32), 3763– 3772.
- Pujade-Lauraine, E., Ledermann, J. A., Selle, F., Gebski, V., Penson, R. T., Oza, A. M., … Friedlander, M. (2017). Olaparib tablets as maintenance therapy. The Lancet Oncology, 18(9), 1274–1284.
- Murai, J., Huang, S. Y. N., Renaud, A., Zhang, Y., Ji, J., Takeda, S., … Pommier, Y. (2014). Synthesis of PARP inhibitors and PARP trapping. Molecular Cancer Therapeutics, 13(2), 433–443.
- Hopkins, T. A., Shi, Y., Rodriguez, L. E., Solomon, L. R., Donawho, C. K., DiGiammarino, E. L., … Luo, Y. (2017). Mechanistic dissection of PARP1 trapping and cytotoxicity. Molecular Cancer Research, 13(11), 1465–1477.
- Shen, Y., Rehman, F. L., Feng, Y., Boshuizen, J., Bajrami, I., Elliott, R., … Lord, C. J. (2020). BMN-673 (talazoparib). Clinical Cancer Research, 26(18), 4568–4579.
- Konstantinopoulos, P. A., Waggoner, S., Vidal, G. A., Mita, M., Moroney, J. W., Holloway, R., … Matulonis, U. A. (2023). Single-arm phases of PARP-immunotherapy combinations. Clinical Cancer Research, 29(4), 771–780.
- Hartwell, L. H., Szankasi, P., Roberts, C. J., Murray, A. W., & Friend, S. H. (1997). Integrating genetic approaches into the discovery of anticancer drugs. Science, 278(5340), 1064–1068.
- O’Neil, N. J., Bailey, M. L., & Hieter, P. (2017). Synthetic lethality and cancer. Nature Reviews Genetics, 18(10), 613–623.
- Cruz, C., Castroviejo-Bermejo, M., Gutiérrez-Enríquez, S., Llop-Guevara, A., Ibrahim, Y. H., Gris-Oliver, A., … Balmaña, J. (2018). RAD51 foci as a functional biomarker of homologous recombination repair. Nature Communications, 9, 4512.
- Lord, C. J., & Ashworth, A. (2023). Mechanisms of resistance to therapies targeting DNA repair. Nature Reviews Cancer, 23(1), 1–17.
- Kondrashova, O., Nguyen, M., Shield-Artin, K., Tinker, A. V., Teng, N. N., Harrell, M. I., … Swisher, E. M. (2017). Secondary somatic mutations restoring BRCA1/2 predict resistance. Journal of Clinical Oncology, 35(11), 1304–1310.
- Ashworth, A. (2008). A synthetic lethal therapeutic approach: Poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. Journal of Clinical Oncology, 26(22), 3785–3790.
- Fong, P. C., Boss, D. S., Yap, T. A., Tutt, A., Wu, P., Mergui-Roelvink, M., … & de Bono, J. S. (2009). Inhibition of poly (ADP-ribose) polymerase in tumors from BRCA mutation carriers. New England Journal of Medicine, 361(2), 123–134.
- Tutt, A., Robson, M., Garber, J. E., Domchek, S. M., Audeh,M. W., Weitzel, J. N., … & Carmichael, J. (2010). Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: A proof-of-concept trial. The Lancet, 376(9737), 235–244.
- Ledermann, J., Harter, P., Gourley, C., Friedlander, M., Vergote, I., Rustin, G., … Pujade-Lauraine, E. (2012). Olaparib maintenance therapy in platinum-sensitive ovarian cancer. New England Journal of Medicine, 366(15), 1382–1392.
- Swisher, E. M., Lin, K. K., Oza, A. M., Scott, C. L., Giordano, H., Sun, J., … Kaye, S. B. (2017). Rucaparib in relapsed ovarian cancer. The Lancet Oncology, 18(1), 75–87.
- Konstantinopoulos, P. A., Ceccaldi, R., Shapiro, G. I., & D’Andrea, A. D. (2015). Homologous recombination deficiency. Cancer Discovery, 5(11), 1137–1154.
- Swisher, E. M., Kwan, T. T., Oza, A. M., Tinker, A. V., Ray-Coquard, I., Oaknin, A., … Kaye, S. B. (2018). Molecular mechanisms of resistance to PARP inhibitors.Journal of Clinical Oncology, 36(15), 1630–1639.
- Parker, J. S., Mullins, M., Cheang, M. C., Leung, S., Voduc, D., Vickery, T., ... & Bernard, P. S. (2009). Supervised risk predictor of breast cancer based on intrinsic subtypes. Journal of clinical oncology, 27(8), 1160-1167.
- Telli, M. L., Timms, K. M., Reid, J., Hennessy, B., Mills,G. B., Jensen, K. C., … Ford, J. M. (2016). Homologous recombination deficiency (HRD) score predicts response to platinum-containing neoadjuvant chemotherapy in patients with triple-negative breast cancer. Clinical Cancer Research, 22(15), 3764–3773.
- Timms, K. M., Abkevich, V., Hughes, E., Neff, C., Reid, J., Morris, B., … Hartman, A. R. (2014). Association of BRCA1/2 defects with genomic scores. Clinical Cancer Research, 20(19), 4879–4888.
- Coleman, R. L., Fleming, G. F., Brady, M. F., Swisher, E. M., Steffensen, K. D., Friedlander, M., … Matulonis, U. A. (2017). Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3). The Lancet, 390(10106), 1949–1961.
- Lheureux, S., Braunstein, M., & Oza, A. M. (2020). Evolving role of PARP inhibitors. Journal of Clinical Oncology, 38(30), 3466–3475.
- Zimmer, A. S., Nichols, E., Cimino-Mathews, A., Peer, C. J., Cao, L., Lee, M. J., … Nanda, R. (2022).A phase I study of olaparib and durvalumab in patients with recurrent ovarian cancer. Clinical Cancer Research, 28(1), 58–67.
- Faraoni, I., & Graziani, G. (2018). Role of BRCA mutations in cancer treatment with PARP inhibitors. Cancers, 10(12), 487.
- Ledermann, J., Harter, P., Gourley, C., Friedlander, M., Vergote, I., Rustin, G., … Pujade-Lauraine, E. (2014). Overall survival in patients with platinum-sensitive ovarian cancer receiving olaparib. The Lancet Oncology, 15(8), 852–861.
- Jenkins, M., Bian, C., & Smith, R. (2018). Pharmacokinetics and safety of niraparib. Clinical Pharmacokinetics, 57(9), 1125–1138.
- Berek, J. S., Matulonis, U. A., Peen, U., Ghatage, P., Mahner, S., Redondo, A., … Mirza, M. R. (2021). Safety and dose modification for patients receiving niraparib. Annals of Oncology, 32(1), 70–78.
- Karlan, B. Y., Hanjani, P., Lin, K., & Monk, B. J. (2020). Individualized dosing of niraparib. Gynecologic Oncology, 156(3), 603–609.
- Oza, A. M., Tinker, A. V., Oaknin, A., Shapira-Frommer, R.,McNeish, I. A., Swisher, E. M., … Domchek, S. M. (2017). Antitumor activity and safety of rucaparib. The Lancet Oncology, 18(1), 75–87.
- Litton, J. K., Rugo, H. S., Ettl, J., Hurvitz, S. A., Gonçalves, A., Lee, K. H., … Blum, J. L. (2018). Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. New England Journal of Medicine, 379(8), 753–763.
- Shen, J., Zhao, W., Ju, Z., Wang, L., Peng, Y., Labrie, M.,… Peng, G. (2019). PARPi triggers DNA damage response.Cancer Research, 79(2), 367–380.
- Karzai, F. H., VanderWeele, D. J., Madan, R. A., Owens, H., Cordes, L. M., Hankin, A., … Gulley, J. L. (2020). Activity of the PARP inhibitor talazoparib in metastatic prostate cancer. Journal of Clinical Oncology, 38(11), 1209–1218.
- O’Connor, M. J., Targeting the DNA damage response. Cancer Cell, 39(2), 173–185.
- Polak, P., Kim, J., Braunstein, L. Z., Karlic, R., Haradhavala, N. J., Tiao, G., … Getz, G. (2017).A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nature Genetics, 49(10), 1476–1486.
- Kim, H., George, E., Ragland, R., Rafail, S., Zhang, R., Krepler, C., … Konstantinopoulos, P. A. (2021). Targeting homologous recombination deficiency in ovarian cancer. Nature Reviews Cancer, 21(10), 645–661.
- Batalini, F., Madison, R. W., Sokol, E. S., Jin, D. X., Chen,K. T., Decker, B., … Ross, J. S. (2022). Homologous recombination deficiency landscape of breast cancers and implications for PARP inhibitor sensitivity. Cancer Research, 82(3), 493–505.
- van Wijk, L. M., Vermeulen, S., Meijers, M., Schouten, P. C., & Jonkers, J. (2022). RAD51 functional assays. Nature Cancer, 3(1), 49–62.
- Coleman, R. L., Oza, A. M., Lorusso, D., Aghajanian, C., Oaknin, A., Dean, A., … Monk, B. J. (2020). Rucaparib maintenance treatment for recurrent ovarian cancer after response to platinum therapy. Journal of Clinical Oncology, 38(15), 150–159.
- Zhou, Z., Dang, Q., Jin, W., Li, X., & Zhang, Y. (2025).Combination strategies with PARP inhibitors: Overcoming resistance and expanding therapeutic benefit. Nature Reviews Clinical Oncology, 22(1), 45–62.
- Ray-Coquard, I., Pautier, P., Pignata, S., Pérol, D., González-Martín, A., Berger, R., … Ledermann, J. A. (2019). Olaparib plus bevacizumab as first-line maintenance therapy. New England Journal of Medicine, 381(25), 2416–2428.
- LaFargue, C. J., Dal Molin, G. Z., Sood, A. K., & Coleman, R.L. (2019). Exploring and comparing adverse events between PARP inhibitors. The Lancet Oncology, 20(1), e15–e28.
- Domchek, S. M., Postel-Vinay, S., Im, S. A., Park, Y. H., Delord, J. P., Italiano, A., … Robson, M. (2020). Olaparib and durvalumab in patients with germline BRCA-mutated metastatic breast cancer (MEDIOLA). Journal of Clinical Oncology, 38(11), 1115–1125.
- Banerjee, S., Moore, K. N., Colombo, N., Scambia, G., Kim,G., Oaknin, A., … & Oza, A. M. (2020). Maintenance olaparib for patients with newly diagnosed advanced ovarian cancer and a BRCA mutation (SOLO1/GOG 3004): 5-year follow-up of a randomized, double-blind, placebo-controlled, phase 3 trial. The Lancet Oncology, 21(1), 82–94.
- Vinayak, S., Tolaney, S. M., Schwartzberg, L., Mita, M., McCann, G., Tan, A. R., … Domchek, S. M. (2019). TOPACIO/KEYNOTE-162: Niraparib plus pembrolizumab in patients with advanced triple-negative breast cancer or recurrent ovarian cancer. JAMA Oncology, 5(8), 1132–1140.
- Wang, Z., Sun, X., Bao, Y., Mo, J., Du, H., Hu, J., & Zhang,X. (2020). Epigenetic regulation in DNA repair. Frontiers in Genetics, 11, 554.
- Mills, J. D., Iyer, A. M., van Scheppingen, J., Bongaarts, A., Anink, J. J., Janssen, B., … Aronica, E. (2020). DNA methylation profiling reveals novel epigenetic signatures in drug-resistant epilepsy. Nature Communications, 11, 5823.
- Sun, H., Zhou, L., Li, J., & Wang, Y. (2018). Multi-omics integration in cancer biology. Briefings in Bioinformatics, 19(5), 1070–1083.
- Cao, C., Zhou, Y., Zhang, H., & Zhang, J. (2023). Homologous recombination deficiency and PARP inhibitors in cancer therapy: Mechanisms and clinical perspectives. Frontiers in Oncology, 13, 1134567.
- Coleman, R. L., Fleming, G. F., Brady, M. F., Swisher, E.M., Steffensen, K. D., Friedlander, M., … & Matulonis, U.A. (2019). Veliparib with first-line chemotherapy and as maintenance therapy in ovarian cancer. New England Journal of Medicine, 381(25), 2403–2415.
- Gelmon, K. A., Tischkowitz, M., Mackay, H., Swenerton, K., Robidoux, A., Tonkin, K., … & Oza, A. (2011). Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: A phase 2 multicentre trial. The Lancet Oncology, 12(9), 852–861.
- McCabe, N., Turner, N. C., Lord, C. J., Kluzek, K., Bialkowska, A., Swift, S., … & Ashworth, A. (2006). Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly (ADP-ribose) polymerase inhibition. Cancer Research, 66(16), 8109–8115.
- New England Journal of Medicine, 379(26), 2495–2505.
- Postel-Vinay, S., Vanhecke, E., Olaussen, K. A., Lord, C. J., Ashworth, A., Soria, J. C., & Andre, F. (2013). The potential of exploiting DNA-repair defects for optimizing lung cancer treatment. Nature Reviews Clinical Oncology, 10(6), 348–357.
- Ramakrishnan Geethakumari, P., Shukla, S., & Ganesan, S. (2023). Mechanisms of resistance to PARP inhibitors—recent advances and future directions. Cancers, 15(7), 2035.
- Shen, Y., Rehman, F. L., Feng, Y., Boshuizen, J., Bajrami, I., Elliott, R., … & Lord, C. J. (2013). BMN 673, a novel and highly potent PARP1/2 inhibitor for the treatment of human cancers with DNA repair deficiency. Clinical Cancer Research, 19(18), 5003–5015.
- Zhou, J., Wang, J., Wang, Z., Li, X., & Xu, Y. (2022). Clinical development of PARP inhibitors in cancer therapy: Progress and challenges. Biochimica et Biophysica Acta (BBA) -Reviews on Cancer, 1877(1), 188676.