
Journal of Ophthalmology & Clinical Research(JOCR)
ISSN: 2573-9573 | DOI: 10.33140/JOCR
Impact Factor: 1.3


Case Report - (2024) Volume 8, Issue 2
Rare Autoimmune Disease of Neonate Diagnosed with The Help of Ophthalmology
Received Date: Aug 19, 2024 / Accepted Date: Sep 25, 2024 / Published Date: Sep 27, 2024
Copyright: ©©2024 Vedanshi Pandya. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Citation: Pandya, V., Nagrecha, P., Vora, N. (2024). Rare Autoimmune Disease of Neonate diagnosed with the help of ophthalmology. J Ophthalmol Clin Res, 8(2), 01-02.
Abstract
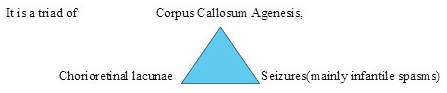
To present an interesting case of “Ophthalmic features in Aicardi Syndrome” Aicardi syndrome is a genetic X-linked defect commonly seen in girls. The ocular finding in Aicardi Syndrome are important because they may be the initial manifestation of the disease.
Objective

To present an interesting case of “Ophthalmic features in Aicardi Syndrome” Aicardi syndrome is a genetic X-linked defect commonly seen in girls. The ocular finding in Aicardi Syndrome are important because they may be the initial manifestation of the disease.
Case Report
A 45 days old Female presented to the NICU with microcephaly, microphthalmos and recurrent seizures. She had a full-term Caesarean delivery. The infant also had missing 7-9 ribs, mild scoliosis and iris coloboma. There was no family history of such condition.
Discussion
Hypothetically Aicardi syndrome occurs due to spontaneous mutation at Xp22 chromosome. Though no exact etiology describes the condition. Although cases occur throughout the world; exact incidence and prevalence is unknown. The syndrome occurs in patients of diverse clinical backgrounds and no racial predominance is noted. Aicardi syndrome is complicated by severe mental retardation, intractable epilepsy and a resultant propensity to pulmonary complications. The condition often leads to death in the first decade. Sudden unexplained death is common. The oldest reported case was 32 years old. Average age of death in west is 8.8 years.
Clinical Features
Hypothetically Aicardi syndrome occurs due to spontaneous mutation at Xp22 chromosome. Though no exact etiology describes the condition. Although cases occur throughout the world; exact incidence and prevalence is unknown. The syndrome occurs in patients of diverse clinical backgrounds and no racial predominance is noted. Aicardi syndrome is complicated by severe mental retardation, intractable epilepsy and a resultant propensity to pulmonary complications. The condition often leads to death in the first decade. Sudden unexplained death is common. The oldest reported case was 32 years old. Average age of death in west is 8.8 years.
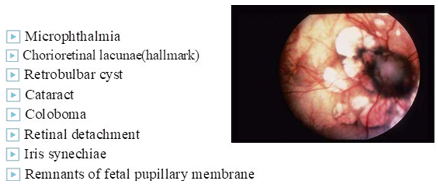
Ophthalmic Features
Investigations
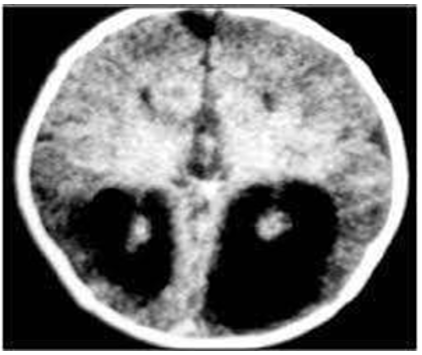
Plain radiographs can be done for skeletal malformations. Based on clinical suspicion, ultrasonography through anterior fontanelle is initial investigation of choice for infants. MRI- brain with spine is gold standard investigation and is preferred. CT scan may be a helpful additional study. EEG- can be done for typical presence of burst suppression pattern, with complete asynchrony between two hemispheres.
Treatment
1. There is no permanent cure.
2. Seizures are treated with multiple anti-epileptic medications. Most effective treatment includes-ketogenic diet, vigabatrin, lamotrigine and topiramate.
3. Patients may benefit from specialized care with physical medicine and rehabilitation specialist as well as physical, occupational and speech therapists.
References
- Salmon, J. F. (2019). Kanski's clinical ophthalmology e-book: a systematic approach. Elsevier Health Sciences.
- Edition;2020;450-453
- Choudhary, M. M., Hajj-Ali, R. A., & Lowder, C. Y. (2014). Gender and ocular manifestations of connective tissue diseases and systemic vasculitides. Journal of ophthalmology, 2014(1), 403042.
- Chaudhuri, Z., & Vanathi, M. (2011). Postgraduate Ophthalmology, Two Volume Set (Vol. 1). Wife Goes On.
- Soheilian, M., Bagheri, A., & Aletaha, M. (2002). Dacryoadenitis as the earliest presenting manifestation of systemic Wegener's granulomatosis.