
Toxicology and Applied Pharmacology Insights(TAPI)
ISSN: 2641-0451 | DOI: 10.33140/TAPI

Research Article - (2023) Volume 6, Issue 1
Virtual Screening on Protein-Ligand Interactions: A Pharmacological Aspect of Zingiber Officinale for COVID-19 Remedy
Received Date: Oct 02, 2023 / Accepted Date: Oct 23, 2023 / Published Date: Nov 23, 2023
Copyright: ©Ã?©2023 Mikidadi S. Gurisha, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Citation: Gurisha, M. S., Makoba, A. (2023). Virtual Screening on Protein-Ligand Interactions: A Pharmacological Aspect of Zin-giber Officinale for COVID-19 Remedy. Toxi App Phar Insig, 6(1), 150-158.
Abstract
With the current pandemic of the novel coronavirus disease 2019(COVID-19) in hand, researchers around the world are dexterously working to find the best suitable drug candidates and to overcome vaccination-related challenges. In Tanzania, ginger (Zingiber officinale) has been taken as a traditional remedy for COVID-19 by processing it into a different drinks. Computer-aided drug discovery provides a promising attempt to allow scientists to develop new and target-specific drugs to fight any disease. Therefore, in this study, Virtual Screening was conducted on 113 phytochemicals derived from the Zingiber officinale herb to find lead molecules for SARS-CoV-2. A total of 10 phytochemicals qualified from PyRx Virtual Screening, out of which only 5, namely Gingerenone A, Jyperin, Meletin, Isorhamnetin and Shogaol demonstrated a substantial binding affinity with D614G SARS-CoV-2 protein, this finding implied that several natural compounds of plants evaluated in this study showed better binding free energy compared to remdesivir which so far are recommended by the FDA in the treatment of COVID-19. Molecular docking analysis was conducted using BIOVIA Discovery Studio, where 7BNO Open conformation of D614G SARS-CoV-2 was used as the protein receptor. Therefore, the present study identifies potential inhibitors of D614G SARS-CoV-2 protein for COVID 19 which needs to be validated further, both experimentally and clinically.
Keywords
Protein-Ligand, COVID-19, SARS-CoV-2 and Zingiber Officinale
Introduction
Coronavirus disease (COVID-19) is an infectious disease caused by the SARS-CoV-2 virus. The pandemic has become the great- est global public health crisis in recent years, and the epidemic is still continuing (WHO, 2022). Due to different levels of social and economic development and health care infrastructure in various regions, not all countries can adopt current medical technologies and obtain sufficient vaccines and therapeutic drugs. Therefore, in some regions, such as Africa, herbal medicine plays an important role in the fight against COVID-19 [1].
Tanzania is one of the African countries that relished the use of traditional medicine as a remedy for the pandemic. The country with a long history of the use of herbs utilizes Ginger, Lemon and Garlic among others for COVID-19 treatment. However, their effi- cacy and safety might be looked into by pharmaceutical industries and researchers.
Recent developments in comprehensive analytical techniques, such as transcriptomics and proteomics, have enabled us to identi- fy proteins associated with active COVID-19 in humans (Preman et al., 2022). SARS-CoV-2 (Severe Acute Respiratory Syndrome Coronavirus 2) also known as 2019-nCoV (2019 Novel Corona- virus) is a virus that causes illnesses ranging from common cold symptoms to severe effects including death and respiratory dis- tress. The CoV Spike (S) protein plays the most important role in viral attachment, fusion and entry, and serves as a target for the development of antibodies, entry inhibitors and vaccines (Thermo Fisher, 2022).
Research for discovering effective drugs to treat COVID-19 has been a priority since the outbreak of the disease. The clinical ap- plication of remdesivir has been greatly restricted by the need for intravenous administration, as well as unstable concentrations in plasma and variable antiviral activity in different organelles [2, 3]. Four neutralizing antibodies (bamlanivimab, etesevimab, casiriv- imab, and imdevimab) have been approved by the United States Food and Drug Administration; however, their high cost and need for intravenous administration render them inaccessible to the public [2].
Therefore, effective and inexpensive oral drugs are the priority for the prevention and control of COVID-19, because they can be used after exposure to SARS-CoV-2 or at the first sign of COVID-19. The use of herbal medicines and phytonutrients or nutraceuticals continues to expand rapidly across the world with many people now resorting to these products for the treatment of various health challenges like COVID-19 (WHO, 2004).
Experimentally and clinically, ginger (the rhizome of Zingiber of- ficinale) has exhibited numerous therapeutic activities, including anti-inflammatory, antioxidative, immunomodulatory, antimicro- bial, anti-fungal, anticancer, neuroprotective, antimigraine, hepa- toprotective, hypo-cholesterolemic, cardiovascular protective, respiratory protective, anti-obesity, anti-diabetic, anti-nausea, and anti-emetic effects [4].
Ginger also displays direct anti-viral effects, and can have a pro- tective role against ARDS [5, 6]. Which is the major cause of mor- tality in patients with severe COVID-19. Computer-aided drug discovery methods have played a major role in the development of therapeutically important small molecules for over three decades [7].
Computational approaches have the power to screen hits from a huge database and design novel small molecules. Computer-aided drug discovery and design not only reduce the costs associated with drug discovery by ensuring that the best possible lead com- pound enters animal studies, but it may also reduce the time it takes for a drug to reach the consumer market (Preman et al., 2022). Mo- lecular docking is one such computational methodology, which is a process that predicts the preferred orientation of one molecule to another when bound together to form a stable complex [7].
There is enumerable software available to perform docking studies among which the BIOVIA Discovery Studio Visualizer (2020) and PyRx Virtual Screening (0.9.x) are the best due to their accuracy and user-friendly options. Docking in glide involves various steps such as Ligand preparation, Protein preparation, and receptor grid generation, docking and scoring [8].
In the present study, we aimed to find the antiviral properties of Zingiber Officinale against COVID-19 through in silico analysis. This makes it easier and less time-consuming in screening lead compounds and their targets therefore, could potentially be used as alternatives herb for immunity boost-up against COVID-19.
Materials and Methods
Protein Preparation
The target protein used in this study is 7BNO Open conformation of D614G SARS-CoV-2 spike with 2 Erect RBDs downloaded from the Protein Data Bank (RCSB PDB) website (https://www. rcsb.org/). Since they typically contain only heavy atoms and in- clude co-crystallized ligands, water molecules, heteroatoms, Ac- tosite, and co-factors were removed since the standard structure from the PDB is not ideal for immediate use [9]. Polar hydrogen atoms were added in this study because crystallographic structures usually lack hydrogen atoms [10]. And the protein receptor was then saved in PDBQT format for further analysis (Figure 1). Pro- tein receptor was prepared using BIOVIA Discovery Studio Visu- alization (2021).

Figure 1: 7BNO Open conformation of D614G SARS-CoV-2 (Protein Receptor)
Ligand Preparation
The native SDF format structures of 6-gingerol, 6-shogaol, 6-para- dol, quercetin, zingerone, and gingerenone-A ligands were down- loaded from Zinc Database (Table 1). The ligand structures were then saved in SDF format for further analysis. The docking li- gands were prepared from the PyRx Virtual Screening software where they were inserted via open babel one after another. The Ligands energy were minimized before converting them into PD- PQT format. All ligand in PDPQT format were forwarded to Vina Wizard and Vina Search space were recorded as follows; center X: 199.647, Y: 243.081, Z: 225.8683, Dimension (Angstrom) X: 117.7926, Y: 143.1828, Z: 25.7037. After running the Wizard, the results were saved as comma separate values (CSV). The ligands with the highest affinity were saved as PDB files ready for dock- ing (Table 2). Docking was performed using BIOVIA Discovery Studio (2021).
|
Name |
Zinc DB ID |
Structure |
Molecular Weight |
H-bond Acceptors |
H-Bond Donors |
High Lipophilicity Log P |
|
Gingerol |
1531846 |
|
294.391 |
4 |
2 |
3.234 |
|
6-Gingerol |
3872686 |
|
294.391 |
4 |
2 |
3.234 |
|
Shogaol |
1531865 |
|
276.376 |
3 |
1 |
4.039 |
|
6-Paradol |
1531857 |
|
278.392 |
3 |
1 |
4.263 |
|
Meletin |
3869685 |
|
302.238 |
7 |
3 |
1.988 |
|
Isorhamnetin |
517261 |
|
316.265 |
7 |
3 |
2.291 |






|
Zingerone Glucoside |
13339791 |
|
356.371 |
0 |
0 |
-0.605 |
|
Jyperin |
3973253 |
|
464.379 |
0 |
0 |
-0.539 |
|
Gingerenone A |
1531844 |
|
356.418 |
5 |
2 |
3.806 |
|
Gingerenone B |
13377938 |
|
386.444 |
6 |
2 |
3.814 |




Table 1: List of Ligands downloaded from Zinc Database and saved in SDF format
Software Used
A. BIOVIA Discovery Studio
BIOVIA Discovery Studio (2020) is a comprehensive software suite for analyzing and modeling molecular structures, sequences, and other data of relevance to life science researchers (Discovery Studio 3.5 Tutorial). The product includes functionality for view- ing and editing data along with tools for performing basic data analysis.
PyRx Virtual Screening
PyRx is a Virtual Screening software (0.9.x) for Computational Drug Discovery that can be used to screen libraries of compounds against potential drug targets. PyRx enables Medicinal Chemists to run Virtual Screening from any platform and helps users in ev- ery step of this process - from data preparation to job submission and analysis of the results.
Results and Discussion
Molecular Docking
BIOVIA Discovery Studio software performs the prediction of bound conformation based on free binding energies, which is cal- culated on the basis of the empirical force field. Vina calculates its own grid maps quickly and automatically [11]. It also clusters and ranks the results. Since molecular docking helps to assess the con- formation of ligands within the binding site with a high degree of accuracy, it has been an important method in drug predictions [12]. Out of the 113 PyRx screened ligands, only 10 passed, which were then subjected to molecular docking. After the docking analysis, only 5 phytochemicals out of these 10 showcased a better binding affinity towards the target protein. The binding affinity score re- produces the potential energy change, when the protein and ligand come together (CLC BIO. 2021). This indicates that a highly neg- ative score relates to a strong binding and a less negative or even positive score refers to weak or non-existing binding. Therefore, the ligands with the least binding affinity scores (from -7.6 to -5.9 kcal/mol) were considered for further study (Table 2).
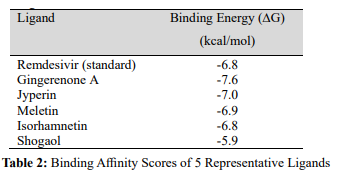
Gingerenone A which is aromatic, pungent enriched with the natu- ral phytochemicals exhibited the least binding affinity score of all the molecules (-7.6 kcal/mol) (Figure 2). Gingerenone A has been detected, but not quantified in, gingers (Zingiber officinale), herbs and spices. This could make gingerenone A a potential biomarker for the consumption of these foods (HMDB. 2022).
Jyperin (-7.0 kcal/mol) is an active compound found in plants of the genera Hypericum and Crataegus, and reported to exhibit anti- oxidant, anticancer, and anti-inflammatory activities [13] (Figure 3).
Melitin, a major bioactive component (-6.9kcal/mol) which can be applied as a means to reduce excessive immune responses and provide new alternatives for the control of inflammatory diseases [14]. Experimental studies show that the biological functions of Melitin could be applied for therapeutic use in vitro and in vivo [15] (Figure 4).
|
Figure No. |
3-D interaction Diagram |
2-D interaction diagram |
|
2 |
A. 3D interaction diagram of Gingerenone A docked with D614G SARS-CoV-2 visualized using Discovery Studio 2020
|
B. 2D interaction diagram of Gingerenone A docked with D614G SARS-CoV-2 visualized using Discovery Studio 2020 |



|
3 |
A. 3D interaction diagram of Jyperin docked with D614G SARS-CoV-2 visualized using Discovery Studio 2020 |
B. 2D interaction diagram of Jyperin docked with D614G SARS-CoV 2 visualized using Discovery Studio 2020 |
|
4 |
A. 3D interaction diagram of Meletin docked with D614G SARS-CoV-2 visualized using Discovery Studio 2020 |
B. 2D interaction diagram of Meletin docked with D614G SARS-CoV 2 visualized using Discovery Studio 2020 |
|
|
A. 3D interaction diagram of Isorhamnetin docked with D614G SARS-CoV-2 visualized using Discovery Studio 2020 |
B. 2D interaction diagram of Isorhamnetin docked with D614G SARS-CoV-2 visualized using Discovery Studio 2020 |






|
6 |
A. 3D interaction diagram of Shogaol docked with D614G SARS-CoV-2 visualized using |
B. 2D interaction diagram of Shogaol A docked with |
|
|
Discovery Studio 2020 |
D614G SARS-CoV 2 visualized using Discovery Studio 2020 |
|
7 |
A. 3D interaction diagram of remdesivir docked with D614G SARS-CoV-2 visualized using Discovery Studio 2020 |
B: 2D interaction diagram of remdesivir docked with D614G SARS-CoV 2 visualized using Discovery Studio 2020 |




Isorhamnetin (-6.8kcal/mol) is one of the most important active in- gredients in the fruits of Hippophae rhamnoides L. and the leaves of Ginkgo biloba L., which possesses extensive pharmacological activities (Figure 5). There have been numerous investigations in the recent years on isorhamnetin, which has the effects of cardio- vascular and cerebrovascular protection, anti-tumor, anti-inflam- matory, anti-oxidation, organ protection and prevention of obesity [13].
Shogaol is reported to be one of the most bioactive components of ginger rhizomes [16] (Figure 6). Shogaol enhances memory and improves the antioxidant defense system. Because of that, it is frequently tested in different neurological disorders including Alzheimer's disease (AD) [17, 18]. It can restore cognitive im- pairment, reduce astrogliosis and microgliosis, and elevate nerve growth factor (NGF) in Aβ and scopolamine-induced mice models of dementia [17].
Compliance with Lipinski's Rule of Five
In drug discovery setting, the Rule of 5 predicts that poor ab- sorption or permeation is more likely when there are more than 5 H-bond donors, 10 H-bond acceptors, the molecular weight which is greater than 500, and the calculated Log P (ILog P) greater than 5 [19]. All selected phytochemicals (Gingerenone A, Jyperin, Meletin, Isorhamnetin and Shogaol) satisfied the condi- tions of Lipinski’s law. Gingerenone A have (2) Hydrogen bond donor, (6) Hydrogen bond acceptor, Molecular Weight (356.418 g/mo) and Log P (3.814). Jyperin have (0) Hydrogen bond donor, (0) Hydrogen bond acceptor, Molecular Weight (464.379 g/mo) and Log P (-0.539). Meletin have (3) Hydrogen bond donor, (7) Hydrogen bond acceptor, Molecular Weight (302.238 g/mo) and Log P (1.988). Isorhamnetin have (3) Hydrogen bond donor, (7) Hydrogen bond acceptor, Molecular Weight (276.376 g/mo) and Log P (1.988). Shogaol have (1) Hydrogen bond donor, (3) Hy- drogen bond acceptor, Molecular Weight (316.265 g/mo) and Log P (4.039). All selected phytochemicals comply with the Lipinski's Law and therefore can be applied and function like drugs.
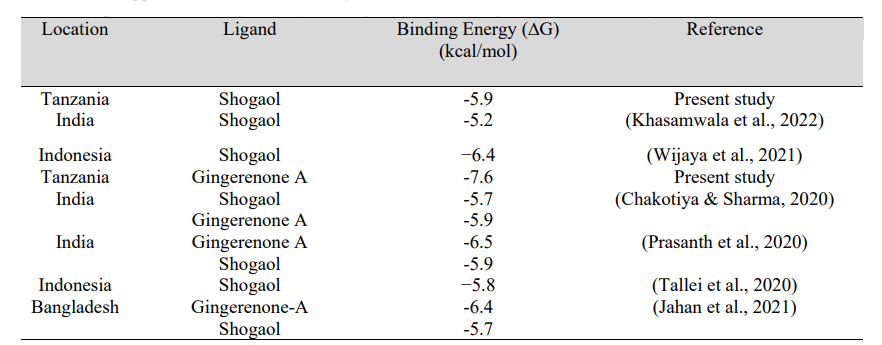
Table 3: Comparison of Binding Affinity of Zingiber officinale ligands from this study with those from other studies
Discussion
The rapid spread of the COVID-19 pandemic has challenged the medical and scientific communities all over the world [21]. De- spite the great technical improvements in drug discovery and me- dicinal chemistry, pharmaceutical development is still expensive and time-consuming (Preman et al., 2022). Using data science to digitally screen chemical libraries to generate drug, leads more efficiently and correctly promises to solve this problem. In sili- co studies are being employed by different researchers to screen natural botanicals based on their classical references for finding suitable lead compounds. In recent research, several attempts have been made to explore their mechanism of action and effects on the host immune system through their activity on target proteins (20) & (Prajapat et al., 2020). Our molecular docking results suggested that the top five compounds all had good binding affinity with the target SARS-CoV-2 protein (Yuan et al., 2022), Gingerenone A, Jyperin, Meletin, Isorhamnetin and Shogaol showed better bind- ing effects among those 10 compounds and also comply with Lip- inski's law. In summary, these compounds can form stable com- plexes with target proteins and were potential active molecules. Therefore, despite the fact that synthetic chemistry dominates the current drug development and manufacturing field, the importance of plant-derived compounds in the treatment and prevention of various diseases cannot be neglected [9]. However, the concrete mechanisms and real effects in COVID-19 treatment still need to explored and confirmed by future work.
Conclusion
Drug development takes long time and it is exhausting process; nevertheless, statistical methods have come to the rescue and have undoubtedly aided in the process's simplification. In the present study, we screened 113 phytochemicals derived from Zingiber officinale herbs to find lead molecules for SARS-CoV-2. A total of 10 phytochemicals qualified from PyRx analysis, out of which only 5, namely Gingerenone A, Jyperin, Meletin, Isorhamnetin and Shogaol exhibited significant binding with D614G SARS-CoV-2 protein. These 5 phytochemicals are present in ginger medicinal available in Tanzania. Thus, the present in silico study identifies potential inhibitors of D614G SARS-CoV-2 protein for COVID 19 which needs to be validated further, both experimentally and clinically.
References
- Liu, Y. X., Zhou, Y. H., Jiang, C. H., Liu, J., & Chen, D. Q. (2022). Prevention, treatment and potential mechanism of herbal medicine for Corona viruses: a review. Bioengineered, 13(3), 5480-5508.
- Fan, H., Lou, F., Fan, J., Li, M., & Tong, Y. (2022). The emer- gence of powerful oral anti-COVID-19 drugs in the post-vac- cine era. The Lancet Microbe, 3(2), e91.
- Schooley, R. T., Carlin, A. F., Beadle, J. R., Valiaeva, N., Zhang, X. Q., et al. (2021). Rethinking remdesivir: synthesis, antiviral activity, and pharmacokinetics of oral lipid prodrugs. Antimicrobial agents and chemotherapy, 65(10), 10-1128.
- Jafarzadeh, A., Jafarzadeh, S., & Nemati, M. (2021). Thera- peutic potential of ginger against COVID-19: Is there enough evidence?. Journal of Traditional Chinese Medical Sciences, 8(4), 267-279.
- Shariatpanahi, Z. V., Mokhtari, M., Taleban, F. A., Alavi, F., Surmaghi, M. H. S., et al. (2013). Effect of enteral feeding with ginger extract in acute respiratory distress syndrome. Journal of critical care, 28(2), 217-e1.
- Xie, X., Sun, S., Zhong, W., Soromou, L. W., Zhou, X., et al. (2014). Zingerone attenuates lipopolysaccharide-induced acute lung injury in mice. International Immunopharmacolo- gy, 19(1), 103-109.
- Sliwoski, G., Kothiwale, S., Meiler, J., & Lowe, E. W. (2014). Computational methods in drug discovery. Pharmacological reviews, 66(1), 334-395.
- Zhang, T., Wei, T., Han, Y., Ma, H., Samieegohar, M., et al.(2016). Protein–ligand interaction detection with a novel method of transient induced molecular electronic spectrosco- py (TIMES): experimental and theoretical studies. ACS cen- tral science, 2(11), 834-842.
- Geethanjali, T., Logesh Kumar, S., Keerthish Sujan, B., Lakshmi Prabhaa, M., Khousikan, K., et al. (2021). Compara- tive Molecular Docking Analysis of Phytoconstituents against Alzheimer’s Disease Targets-An In-Silico Approach. Interna- tional Journal of Research in Pharmaceutical Sciences, 12(2), 1579-1589.
- Jahan, R., Paul, A. K., Bondhon, T. A., Hasan, A., Jannat, K., et al. (2021). Zingiber officinale: Ayurvedic uses of the plant and in silico binding studies of selected phytochemicals with Mpro of SARS-CoV-2. Natural Product Communications, 16(10), 1934578X211031766.
- Trott, O., & Olson, A. J. (2010). AutoDock Vina: improving the speed and accuracy of docking with a new scoring func- tion, efficient optimization, and multithreading. Journal of computational chemistry, 31(2), 455-461.
- Ferreira, L. G., Dos Santos, R. N., Oliva, G., & Andricopulo,A. D. (2015). Molecular docking and structure-based drug de- sign strategies. Molecules, 20(7), 13384-13421.
- Gong, G., Guan, Y. Y., Zhang, Z. L., Rahman, K., Wang, S. J., et al. (2020). Isorhamnetin: A review of pharmacological effects. Biomedicine & Pharmacotherapy, 128, 110301.
- Son, D. J., Kang, J., Kim, T. J., Song, H. S., Sung, K. J., et al. (2007). Melittin, a major bioactive component of bee venom toxin, inhibits PDGF receptor beta-tyrosine phosphorylation and downstream intracellular signal transduction in rat aortic vascular smooth muscle cells. Journal of Toxicology and En- vironmental Health, Part A, 70(15-16), 1350-1355.
- Lee, G., & Bae, H. (2016). Anti-inflammatory applications of melittin, a major component of bee venom: Detailed mech- anism of action and adverse effects. Molecules, 21(5), 616.
- Deb, S., Mazumder, M. K., Dutta, A., Phukan, B. C., Bhat- tacharya, P., et al. (2019). Therapeutic implications of anti-in- flammatory natural products in Alzheimer’s disease. In Dis-covery and Development of Anti-Inflammatory Agents from Natural Products (pp. 241-258). Elsevier.
- Moon, M., Kim, H. G., Choi, J. G., Oh, H., Lee, P. K., et al. (2014). 6-Shogaol, an active constituent of ginger, attenuates neuroinflammation and cognitive deficits in animal models of dementia. Biochemical and biophysical research communica- tions, 449(1), 8-13.
- Shim, S., & Kwon, J. (2012). Effects of [6]-shogaol on cho- linergic signaling in HT22 cells following neuronal damage induced by hydrogen peroxide. Food and chemical toxicolo- gy, 50(5), 1454-1459.
- Benet, L. Z., Hosey, C. M., Ursu, O., & Oprea, T. I. (2016). BDDCS, the Rule of 5 and drugability. Advanced drug deliv- ery reviews, 101, 89-98.
- Khasamwala, R. H., Ranjani, S., Nivetha, S. S., & Hemalatha,S. (2022). COVID-19: An in silico analysis on potential ther- apeutic uses of Trikadu as immune system boosters. Applied biochemistry and biotechnology, 1-11.
- Qazi, S., Das, S., Khuntia, B. K., Sharma, V., Sharma, S., et al. (2021). In silico molecular docking and molecular dynamic simulation analysis of phytochemicals from Indian foods as potential inhibitors of SARS-CoV-2 RdRp and 3CLpro. Nat- ural Product Communications, 16(9), 1934578X211031707.
- Chakotiya, A. S., & Sharma, R. K. (2020). Phytoconstituents of zingiber officinale targeting host-viral protein interaction at entry point of sars-COV-2: A molecular docking study. Def. Life Sci. J, 5(4), 268-277.
- Prasanth, D. S. N. B. K., Panda, S. P., Rao, A. L., Chakravarti, G., Teja, N., et al. (2020). In-silico strategies of some selected phytoconstituents from zingiber officinale as sars cov-2 main protease (COVID-19) inhibitors. Indian J Pharm Educ Res, 54, s552-s559.
- Tallei, T. E., Tumilaar, S. G., Niode, N. J., Kepel, B. J., Idroes, R., et al. (2020). Potential of plant bioactive compounds as SARS-CoV-2 main protease (M pro) and spike (S) glycopro- tein inhibitors: a molecular docking study. Scientifica, 2020.