
Toxicology and Applied Pharmacology Insights(TAPI)
ISSN: 2641-0451 | DOI: 10.33140/TAPI

Research Article - (2023) Volume 6, Issue 1
Polycystic Ovary Syndrome (PCOS) Inhibitory Drug Screening from Selective Inositols
2Depertment of Biochemistry and Molecular Biology, Faculty of Life Science, Bangabandhu Sheikh Mujibu, Bangladesh
Received Date: Oct 23, 2023 / Accepted Date: Nov 16, 2023 / Published Date: Nov 23, 2023
Copyright: ©Ã??Ã?©2023 M A Hasan Roni, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Citation: Shohag, S., Akhter, S., Antora, S, T., Nawer, K, A., Alim, A., et al. (2023). Polycystic Ovary Syndrome (PCOS) Inhibitory Drug Screening from Selective Inositols. Toxi App Phar Insig, 6(1), 166-180.
Abstract
Polycystic ovary syndrome (PCOS) is a heterogeneous ailment characterized by a combination of symptoms, including signs of androgen excess (hirsutism and/or hyperandrogenemia), ovarian dysfunction-oligo-ovulation and/or polycystic ovarian morphology (PCOM)), reproductive abnormalities, obesity, menstrual irregularity, type 2 diabetes (T2DM), hypertension, dyslipidemia, and depression. PubChem, enriched with 110 million chemicals, yielded 6327 inositol derivatives. Lipinski rule of five (MW up to 500 g/mol, H-bond donors 1–5, H-bond acceptors up to 10) criteria has been used for filtration of 582 compounds initial stage. Using escalating molecular weight, 101 molecules were selected from 582, to control this and rogen. To explore the possibility of androgen inhibition, various computational approaches, such as PASSprediction,protein-ligand interactions through molecular docking, chemical descriptors by DFT study, ADMET, bioavailability, and molecular dynamic (MD) simulation, were adopted by using the most accepted methods. Out of 101 inositol derivatives, 90 compounds were found to have more testosterone-inhibiting properties than ovulation-inhibiting or insulin-promoting properties in the PASS prediction investigation. Molecular docking analysis reveals a binding propensity for inositols of more than -7.4 kcal/mol between the androgen receptor protein (3RLJ) and inositols. Some of the most frequent bonded sites are shown to be at the MET:745 and ARG:752 amino acid residues, where the ligand (inositols) forms both hydrogen and hydrophobic bonds. In contrast, the Density Functional Theory (DFT) was used to predict the chemical and physical characteristics of promising inositols in this research. Furthermore, ADME, toxicity, and carcinogenicity factors were analyzed to validate the drugs' potential as safe, effective treatments for PCOS.MD simulation was done for the ligands L60, L77, and L88 against the target protein. The result of RMSD and RMSF revealed that the stability of docked complex is very high. As a conclusion, the in silico research suggests that the three inositols (L60, L77, and L88) are the most promising candidates for PCOS-preventative drugs that still need clinical or additional study.
Keywords
PCOS, Inositols, Molecular Docking, DFT, Drug Discovery, Molecular Dynamics
Graphical Abstarct
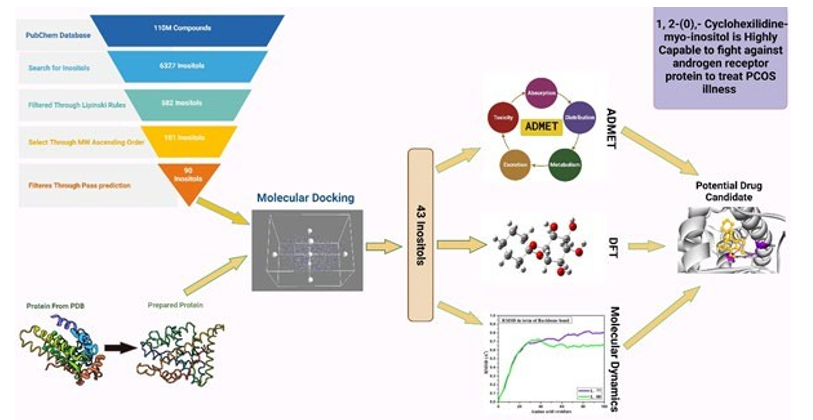
Introduction
Polycystic ovary syndrome (PCOS) is a common endocrine and metabolic disorder that has long-term effects on many parts of a woman's health. Additionally, 6-20% of reproductive- aged women are affected by it (18–44 years) and it lasts well beyond reproductive age [1-3]. A lot of people have faced many problems with their reproductive system due to PCOS, which includes obesity, hyperandrogenism, ovarian dysfunction, irregular menstruation, type 2 diabetes (T2DM), cardiovascular risk factors, for instance, high blood pressure, dyslipidemia, and obstructive sleep apnea (OSA), as well as depression. PCOS- affected pregnant women are at a higher risk of pregnancy complications, such as gestational diabetes and early birth [4-7].
The exact etiology of the polycystic ovarian syndrome is unknown. However, some researchers suggest that polycystic ovarian syndrome is genetically influenced by the prenatal environment, lifestyle, habits, or both. Since there is no potential cure for PCOS, current management strategies are ineffective [8, 9]. Many pharmacological targets may be used to create a novel treatment for PCOS. These terms refer to the "steroid hormone receptor signaling pathway" or "steroid hormone receptor activity" and the "steroid binding" thatoccursas a result of the steroid hormone connection. The imbalance of steroid hormone receptor signaling is considered the main and cardinal cause of PCOS. In general, the steroid hormones are mostly composed of androgens and estrogens. It is simply common fact that women typically have high levels of androgens, which leads to the more affected trend of PCOS. That is why two critical pharmacological targets are concentrated at "androgen receptor signaling pathway" and "androgen receptor binding." As estrogens are required for the proper development and function of the male and female reproductive systems, "estrogen receptor signaling pathway" and "estrogen receptor binding" are two additional significant pharmacological targets [10].
However, the androgen receptor has been considered the cardinal factor to cause PCOS in both male and female bodies, regarding that fact; it is selected for this study to detect the perfect binding agents by Inositol derivatives. In general, androgens possess their steroidal effects through the androgen receptor, which is critical for female reproductive maintenance by promoting follicle development, health, and ovulation [11]. The most frequent and apparent symptom of PCOS, hyperandrogenism, is caused by elevated levels of various androgens, the most common being testosterone. By reducing the hyperandrogenic effect of testosterone, various potential drugs have been explored in this study of PCOS in women [8, 12].
Even though there is no objective test for PCOS, androgen levels in the blood can be checked for hyperandrogenism by using biochemistry. There are many misconceptions about this topic [13]. Various experimental studies have still suggested that inositol medication may have some possible scope in PCOS patients although it has a crucial role in oocyte fertility [14-16]. Inositol treatment is safe and has few side effects compared to other ovulation-induction therapeutic options, such as oral contraceptive therapy (OCT), insulin sensitizer metformin, high testosterone therapy, and clomiphene citrate (CC), and these types of therapies have some risks because metformin and OCT cannot be suggested for treatment during pregnancy due to some complications. Furthermore, their effect on PCOS patients is still unclear [9, 17-20]. The most common used drugs for androgen receptors are cyproterone acetate and spironolactone, which are not estimated to be pure antiandrogens or even anti-steroid receptors, although they may illustrate some drawbacks after being posed in patients bodies [21]. In women with PCOS, 3 months after metformin treatment, the expression of AR was reduced in epithelial and stromal cells in comparison to their levels before treatment [22].
Inositol can be utilized as an efficacious, safe, and successful medication for PCOS, including improved oocyte follicular growth and maturation as well as in IVF stimulation and pregnancy outcomes [23]. Some derivatives of the inositol medicines are now on the market, but they have some drawbacks. Currently, the two most common inositol stereoisomers (my inositol and D-chiro-inositol) being proposed for PCOS treatment have some ratio dependency [24, 25]. Moreover, high D-chiro-inositol dosages can be detrimental to oocytes and fertility [26, 27]. There is a lack of information on the significance of myo-inositol (MI) and d-chiro-inositol (DCI) and there are some research limitations on inositol therapy. Medication containing inositol improves ovarian follicle quality, menstrual cycle quality, and insulin resistance in people with polycystic ovary syndrome. In the future, this medication is predicted to be efficacious in PCOS patients, because it can reduce androgen levels in women [9, 28]. This research has focused on developing a novel drug candidate based on inositol derivatives by using a variety of established computational methods applicable to the treatment of PCOS. The In-silico is truly a sophisticated method of theoretical and computational analysis that is beneficial for identifying new compounds against any disease with its cost effectiveness and consumption of vast labor and time [29]. Calculating a compound's binding capacity in traditional drug discovery and development has considered the primary criteria before going to perform a large-scale in-vitro and in-vivo experiment. However, molecular docking simplifies the procedure in a short amount of time [30]. Pharmacokinetics, toxicity, protein-ligand interaction, chemical reactivity, molecular orbital properties, various optimization methodologies, and MD simulation are all commonly used to discover, develop, and examine drugs and other biologically active molecules using a computer-aided drug design process [31].
The current in-silico investigation has revealed that some inositol derivatives can interact with the androgen receptor with high efficiency. As a consequence; we have concentrated our efforts in this study on using computer-aided drug design to identify potential drug-like compounds targeting the androgen receptor protein.
Methodology
Target Protein Identification, Retrievation, and Preparation
The 3D confirmer of androgen receptor protein used in this study was obtained in PDB format from the RCSB Protein Data Bank (https://www.rcsb.org/) [32]. Prior to obtaining this protein from PDB, this specific PDB identifier-containing single chain mutation-free protein was chosen from UniPort (https://www. uniprot.org/), which was used to look for more specific proteins in the human body. Then, using several criteria such as method, resolution, chain, and position, 3RLJ was chosen from among the various PDB IDs containing protein. The RCSB protein data bank revealed the crystal structure of androgen receptors (3RLJ). It has a molecular mass of 29.21 KD. It is made up of a single amino acid chain with 247 AA sequence lengths and a resolution of 1.90 Å, as determined by the X-ray diffraction method [33]. The crystal structure of 3RLJ protein was prepared using PyMol software (http://www.pymol.org) by eliminating unwanted water and ligand. PyMOL is an open-source molecular graphics tool and has been widely used for 3D visualization of macromolecules [34]. Blind docking was employed using the PyRxautodock tool to experience the binding affinity of the target ligand to the protein. After getting the binding mode of the protein with the ligand, the CASTp server was used to examine the binding site of the protein. From there, pocket 1 was considered the main binding site for having large area coverage and volume (250.505 SA and 110.475 SA). A total of 28 amino acid residues were found in pocket 1, starting from residue number 701 and spanning to 903. From this result, we confirmed that our top three binding affinity-holding ligands bind exactly on the binding site that was predicted through the CASTp server. And these results were presented in the result section of the manuscript at the point “Protein-Ligand Interaction Analysis”.
Retrievation through Pass Prediction
Initially, inositol derivatives were sought in the PubChem database (https://pubchem.ncbi.nlm.nih.gov/), which was enriched with 110 million compounds and yielded 6327 inositol derivatives. From there, 582 compounds were filtered using Lipinski rules (MW up to 500 g/mol, H-bond donor 1–5, H-bond acceptor up to 10). Primarily, 101 compounds were chosen from a total of 582 compounds by using molecular weight in ascending order. The PASS Online server (http://way2drug.com/PassOnline/), which predicts more than 300 pharmacological effects and biochemical mechanisms based on the structural formula of a substance, was used to find new ligands for the required biological targets. Out of these 101 compounds, 90 displayed testosterone-17 beta- dehydrogenase (NADP+) inhibition activity, whereas Pa > Pi and Pa > 0.5.
Ligand preparation and chemical descriptors
The Gaussian 16W software package was used to optimize each of the ligand optimized structures using the density functional theory (DFT), method B3LYP with a 6-31G basis set [35-38]. The main rationale of the Becke, 3-parameter Lee–Yang–Parr (B3LYP) method is that it is widely used for DFT calculations because it is highly capable of giving the accurate results for the electronic structure and quantum calculations. However, it is not dependent on the non-covalent interaction of systems. Due to having the more electronegative atom, like oxygen, the 6-31G basis set was used to get more accurate results for calculating the polarization effect of molecular systems regarding their quantum properties. After optimization, the Gauss View 6.0.16 software was used to viewed and analyze the molecules. The precise estimation of electronic properties, as well as the electron transport potential by evaluating the electrostatic 3D map, was obtained using DFT in this study [39]. The HOMO (highest occupied molecular orbital) and LUMO (lowest unoccupied molecular orbital) are two frontier molecular orbitals that have been used to predict chemical descriptors [40]. To measure molecule stability and reactivity, the EHOMO/ELUMO gap is used. A molecule with a large HOMO-LUMO gap has low stability and, thus, low chemical reactivity. Most reactivity descriptors, such as electrophilicity (ω), chemical potential (μ), electronegativity (x), hardness (η), and softness (S), can also be assessed using some relationship with HOMO and LUMO energia. The ionization energy (Ι) and electron affinity (A) can be determined using I = –HOMO and A = –LUMO values, respectively [41].
The following equations are used in this study:
Energy gap (E gap) = ELUMO - EHOMO… (1)
Chemical Potential (μ) = - (I+A)/2… (2)
Hardness (η) = (I-A)/2 (3)
Softness (σ) = 1/η… (4)
Electronegativity (x) = (I+A)/2… (5)
Electrophilicity (ω) = μ2/2η (6)
Molecular Docking
The virtual screening tool PyRx was used for molecular docking to identify the binding affinity of the desired protein with the selected ligand [42]. For docking purposes, the default configuration parameters of the PyRx virtual screening tools were used. The ligands and protein structures were converted to PDBQT format for docking, with the grid set to X = 20.9047, Y = 5.6066, and Z = 10.2552, and the box dimensions (Å) X = 55.6226973915, Y = 42.5333397961, and Z = 58.0472252083. The grid box parameters were determined to cover the protein's substrate-binding pocket. The binding affinities were calculated in kcal/mol units.
Protein-Ligand Interaction
The molecular interaction of the target ligand with the desired protein aids in our understanding of the ligand's molecular mechanism within the biological system [43]. The post-docking analysis of the non-covalent bond with the bond distance between the protein and ligand was observed through the Biovia Discovery Studio 4.0 client visualizer tool (http://accelrys.com/ products/discovery-studio/). Before this, the docked protein and the desired ligand were bound using PyMol software.
ADMET Property Prediction
The term pharmacokinetics refers to the absorption, distribution, metabolism, and excretion (ADME) processes that are utilized in the computer-aided drug design method to predict the time course of drug movement into in and out of the body. In this investigation, the Swiss ADME (http://www.swissadme.ch/ index.php) database was used to predict various pharmacokinetic parameters such as Molecular Weight, H-bond Donors, H-bond Acceptors, Molecular Formula, Topological Surface Area, TPSA (Polarity), XLOGP3 (Lipophilicity), ESOL (Water Solubility), GI absorption, Bioavailability, and Fraction of sp3 Hybridized Carbon (Csp3) [42, 43].
Toxicity prediction is another crucial part of drug development because it predicts a chemical compound's adverse effects inside the human or animal body before it goes into a clinical trial. The admet SAR server (http://lmmd.ecust.edu.cn/admetsar2/) was used to predict carcinogenicity and acute oral toxicity of selected compounds [44].
Molecular Dynamics Simulation
MD simulation was used to support the docking results for the most effective antiviral drugs. For holo-form, a 100 ns MD simulation was used, (drug-protein). For all simulations, the NAMD dynamics program was used which can be conducted interactively via live view or in batch mode [45]. The top three binding affinity docked complexes of drugs and proteins were docked using the AMBER14 force field, and the MD simulation was used to support those results up to 100 ns for the holo- form (drug-protein). The total system was equilibrated with 0.9 percent NaCl at 298 K in the presence of a water solvent. The atomy Mesh Ewald algorithm for radial electrostatic interactions was discussed through simulation. During the simulation, a cubic cell was propagated within 20Å on each side of the process, and a periodic boundary circumstance was proposed. For the 100 ns MD simulation, a time step of 1.25 fs was used, and snapshots were taken at regular intervals. Analyses of the RMSD and RMSF were performed in VMD once the simulation was completed. After completing the MD simulation, the Ramacharanplot, covariance map, and B-factor/mobility from the molecular dynamic study were calculated and presented as graphical representations.
Results and Discussion
Pass prediction analysis of Target Ligands
The Pass online database was used to assess the overall bioactivities of all compounds based on their chemical structures, as well as to estimate the activity spectrum of the chemical compounds Pa (probability to be active) and Pi (probability to be inactive), both of which have values ranging from 0.000 to 1.000 (expressed as a percentage of probability [46]. Various androgens, the most notable of which is testosterone, are high in women with hyperandrogenic PCOS. So, from the 110 million compounds in the database, a total of 6327 inositol derivatives were discovered to inhibit testosterone. Out of these 6327 Inositols, 587 were filtered using the Lipinski rules. One hundred and one (101) compounds were selected from the 587 inositol mentioned above for this study based on the ascending order of molecular weight of the molecules. Pass prediction was first investigated with these 101 inositol derivatives to identify testosterone inhibition activity. At the same time, the ovulation inhibition activity and insulin promotion activity of these compounds were predicted and compared with testosterone inhibition activity.
Two parameters were selected: (i) Pa > Pi and (ii) Pa > 0.5. Interestingly, of these 101 compounds, 90 compounds revealed testosterone 17 beta-dehydrogenase (NADP+) inhibition activity, and their Pa (probability to be active) value was greater than Pi (probability to be inactive) and Pa > 0.5 as well. Among the three medicinal qualities, they are more responsive to the testosterone inhibition property. Hence, 90 compounds (around 90%) were selected for further study, starting with molecular docking with the androgen receptor protein (3RLJ). Though the selected molecules are testosterone responsive, they would be highly responsive to androgen receptor protein by default. After completing the molecular docking study, 47% of molecules (43 out of 90) showed drug-liking properties based on the docking score. It indicates the better outcome of this prediction study in drug screening than any other large data set. This represented data set (S1) may be used for ovulation inhibition and insulin promotion-related drug discovery research in any other study. Table1 represents the pass prediction (Pa and Pi value) of the 10 best compounds with molecular weight (g/mol) and PubChem CID.
|
Ligand No / Drug |
PubChem CID |
Molecular Weight (g/mol) |
Pa>Pi |
Docking Score (Binding Affinity) Kcal/mol |
|
L-77 |
10858206 |
260.28 |
0.855>0.013 |
-7.5 |
|
L-88 |
102130598 |
260.28 |
0.815>0.022 |
-6.8 |
|
L-60 |
102460862 |
244.28 |
0.848>0.015 |
-6.8 |
|
L-30 |
14757245 |
220.22 |
0.815>0.022 |
-6.6 |
|
L-5 |
101701253 |
204.18 |
0.882>0.009 |
-6.6 |
|
L-67 |
101864951 |
248.27 |
0.804>0.024 |
-6.6 |
|
L-47 |
102258382 |
238.19 |
0.841>0.016 |
-6.6 |
|
L-33 |
58877648 |
222.19 |
0.911>0.005 |
-6.5 |
|
L-34 |
122218899 |
222.19 |
0.911>0.005 |
-6.5 |
|
L-89 |
126968731 |
260.28 |
0.890>0.008 |
-6.5 |
|
Metformin (D1) |
4091 |
129.16 |
0.490>0.063 |
-5.2 |
|
Spironolactone (D2) |
5833 |
416.6 |
0.670>0.008 |
-8 |
|
Cyproterone acetate (D3) |
9880 |
416.9 |
0.569>0.026 |
-7.7 |
Table1: Ligand No, PubChem CID, Molecular Weight, and Pa>Pi value of top 10 molecules based on Docking Score (Binding Affinity).
Molecular Docking Analysis
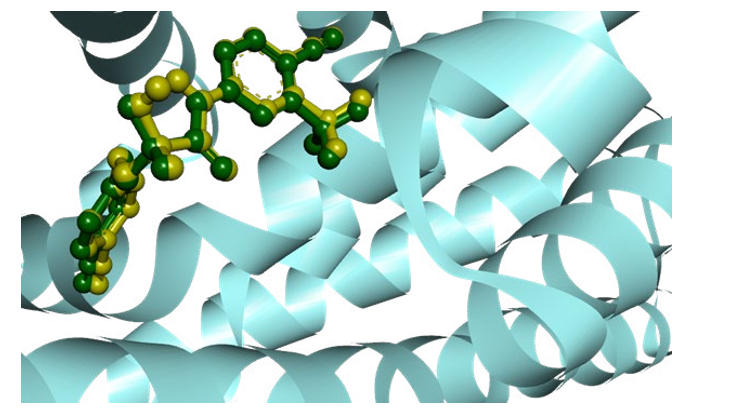
Figure1: Auto dock Vina's predicted pose after molecular docking for the validation of the docking protocol.
Here, the stick representations of ligands denote the superimposed view of docked (magenta) and co-crystallized ligand (green). One of the most important techniques in computer-aided drug design is molecular docking. Its objective is to evaluate the best ligand-binding mode match to a macromolecular partner (proteins), and it typically involves generating several potential ligand conformations within the protein binding site, allowing for the best intermolecular interaction between the desired protein and the target compound [47]. In order to ensure the accuracy of the docking program, the enobosarm crystal configuration was re-docked into the binding site of the androgen receptor protein (3RLJ) using particular docking parameters and scoring systems. For the ligand to attach to the active site cleft, PyMol was used to superimpose the newly re-docked complex onto the reference co-crystallized complex (Figure 1). As shown in (S2), the binding affinities of the target compounds discovered after molecular docking ranged from -4.3 to -7.5 kcal/mol afterward docking protocol validation. The binding affinities of the target compounds discovered after molecular docking ranged from -4.3 to -7.5 kcal/mol, as shown in (S2). Out of 90 docked molecules, the top 43 compounds were chosen for further study, which have a greater binding affinity ranging from -6.0 to -7.5 kcal/mol.
Out of the top-ranked 43 compounds, 33 are suitable drug candidates with docking scores ranging from -6.0 to -6.4, and another 10are potential and promising drug candidates. The docking score with the target androgen receptor protein (3RLJ) ranges from -6.5 to -7.5 kcal/mol. All 10 potential drug candidates' binding affinity is depicted in Table1. Among the very potential 10 drug candidates, the top three are 1,2-O-Cyclohexylidene- myo-inositol (L77), 1-O,4-O-Diallyl-D-myo-inositol (L88), and 1-O,2-O:3-O,4-O-Diisopropylidene-5-deoxy-L-chiro-inositol (L60), which binding scores are -7.5, -6.8, and -6.8 kcal/mol respectively. These three compounds, out of 43, may have a high potential for treating PCOS.
|
Androgen Receptor Protein (3RLJ) |
Androgen Receptor Protein (3RLJ) |
||||||||
|
|
Hydrogen bond |
Hydrophobic bond |
|
Hydrogen bond |
Hydrophobic bond |
||||
|
L/N |
Interacting residue of amino acid |
Distance (Å) |
Interacting residue of amino acid |
Distance (Å) |
L/N |
Interacting residue of amino acid |
Distance (Å) |
Interacting residue of amino acid |
Distance (Å) |
|
L77 |
A:GLN:711 A:MET:745 |
2.42 2.78 |
Absent |
Absent |
L67 |
Absent |
Absent |
A:GLN:711 A:MET:895 A:LEU:701 A:LEU:704 A:MET:780 |
3.43 5.02 5.32 4.01 4.94 |
|
L88 |
A:ARG:752 A:GLU:681 A:GLN:711 A:GLN:711 A:LYS:808 |
2.50 2.67 3.03 2.88 2.85 |
A:ARG:752 A:GLY:683 A:VAL:684 A:PRO:766 A:VAL:715 A:LEU:744 A:LYS:808 A:TRP:718 A:TRP:718 |
3.60 3.33 4.10 4.60 4.56 3.64 4.03 4.65 3.60 |
L47 |
A:GLU:681 A:LYS:808 |
2.35 2.16 |
A:TRP:718 A:VAL:715 A:LEU:744 A:LYS:808 A:TRP:718 |
3.69 4.40 3.80 4.22 4.65 |
|
L60 |
A:ARG:752 A:GLY:683 |
2.12 2.35 |
A:PRO:682 A:ALA:748 A:ALA:748 A:ALA:748 A:ARG:752 A:ARG:752 A:PRO:682 A:VAL:715 A:LEU:744 A:LYS:808 A:TRP:718 A:TRP:718 A:PHE:804 |
4.04 4.46 3.43 3.67 5.07 4.48 3.98 4.04 4.87 4.15 4.33 4.33 5.33 |
L33 |
A:PRO:682 A:GLN:711 A:LYS:808 |
2.14 2.30 2.67 |
Absent |
Absent |
|
L30 |
A:LYS:808 A:GLU:681 A:GLU:681 A:GLY:683 |
2.64 2.52 1.90 2.15 |
A:VAL:715 A:LEU:744 A:LYS:808 A:TRP:718 A:TRP:718 |
4.24 4.21 4.51 4.41 3.65 |
L34 |
A:LYS:808 A:GLU:681 A:PRO:682 A:ARG:752 A:GLY:683 |
2.66 1.83 2.76 2.72 2.67 |
Absent |
Absent |
|
L5 |
A:GLU:681 A:ARG:752 |
2.63 2.60 |
Absent |
Absent |
L89 |
A:LYS:822 A:ASP:732 A:GLN:902 A:GLN:902 |
2.45 1.97 2.36 2.92 |
A:PRO:817 |
4.22 |
|
D1 |
A:GLU681 A:TRP718 |
2.61 2.78 |
Absent |
Absent |
D2 |
A:ARG752 |
2.13 |
Absent |
Absent |
|
D3 |
A:ASN756 |
1.97 |
A:PRO801 |
4.04 |
|
|
|
|
|
Table2: Protein-Ligand interactions of Androgen receptor protein (3RLJ) with top 10 Inositols.
Protein-Ligand Interaction Analysis In this study, top-ranked 43 compounds were chosen for further investigations, all of which had a-6 or higher negative binding affinity with desired protein, and all 43 compounds are capable of being drugs. Biovia Discovery Studio was used to examine the protein-ligand interactions of these 43 compounds that contained hydrogen bonds and hydrophobic bonds (non- bond interactions) with varying bond distances. Protein-ligand Interaction and identification of binding residues are depicted in S3. CASTp (http://sts.bioe.uic.edu/castp/index.html?4jii) server was used to predict the binding pocket and active binding residues, where a total of 27 pockets were found in the protein, tabulated in (S4) and pocket 1 was considered the main binding site for having large area coverage and volume (250.505 SA and 110.475 SA). A total of 28 amino acid residues were found in pocket 1, starting from residue number 701 and spanning to residue number 903 (S5). During molecular docking, the ligands were permitted to move around freely over the protein surface in order to bind to the protein's binding cleft that was most appropriate. A comparison of docked ligands' binding sites with the CASTp active site model was performed after molecular docking in order to make a more accurate drug prediction.
Interaction of Reference Drug with Target protein is depicted in Figure 2
Figure 2: Interaction of Reference Drug with Target protein
A total of 47 binding sites were observed among these 43 molecules where these ligands can bind to treat PCOS illness. Among these 47 binding sites, A: GLN: 711, A:MET:745, and A:ARG:752, were the most common. The lowest bond distance was observed for the binding site A: GLU:681, with a bond distance of 1.83 Å. The top 10 ligands (L77, L88, L60, L30, L5, L67, L47, L33, L34, and L89) binding interactions with the androgen receptor protein (3RLJ) with bond distance are depicted in Table2. The top three binding affinity values were -7.5, -6.8, and -6.8 kcal/mol for the compounds, namely 1,2-O-Cyclohexylidene- myo-inositol (L77), 1-O,4-O-Diallyl-D-myo-inositol (L88), and 1-O,2-O:3-O,4-O-Diisopropylidene-5-deoxy-L-chiro-inositol (L60), and their binding interaction was interpreted to show substantial hydrogen and hydrophobic bonding between the protein and the ligand which were better than reference drugs.
Figure 3: Interaction of the best three moleculeswith 3RLJ protein
For the compound L77, it has been found to form two hydrogen bonds at the positions of A:GLN:711 (2.42 Å), A:MET:745 (2.78 Å) with the desired androgen receptor protein (3RLJ) depicted in Figure 3. Here both the binding sites are active binding sites found in pocket1.
In the case of the compound L88, it has been observed to form five hydrogen bonds and nine hydrophobic bonds with the desired protein. Five hydrogen bonds have been noticed at the position of A:ARG:752 (2.50 Å), A:GLU:681 (2.67 Å), A:GLN:711 (3.03 Å), A:GLN:711 (2.88 Å), A:LYS:808 (2.85 Å) where nine Hydrophobic bonds have found to form at the position of A:ARG:752 (3.60 Å), A:GLY:683 (3.33 Å), A:VAL:684 (4.10 Å), A:PRO:766 (4.60 Å), A:VAL:715 (4.56 Å), A:LEU:744 (3.64 Å), A:LYS:808 (4.03 Å), A:TRP:718 (4.65 Å), A:TRP:718 (3.60 Å) showed in Figure 3.Here, A:ARG:752, an active binding site from the pocket 1form both hydrogen and hydrophobic bonds, and A:GLN:711 formsan only hydrogen bonds.
The interaction study of the compound L60 was determined that found two hydrogen bonds at the position of A:ARG:752 (2.12 Å), A:GLY:683 (2.35 Å) and thirteen hydrophobic bonds at A:PRO:682 (4.04 Å), A:ALA:748 (4.46 Å), A:ALA:748 (3.43 Å), A:ALA:748 (3.67 Å), A:ARG:752 (5.07 Å), A:ARG:752 (4.48 Å), A:PRO:682 (3.98 Å), A:VAL:715 (4.04 Å), A:LEU:744 (4.87 Å), A:LYS:808 (4.15 Å), A:TRP:718 (4.33 Å), A:TRP:718 (4.33 Å), A:PHE:804 (5.33 Å) residual position depicted in Figure 3.Here the active binding site from pocket 1 isA:ARG:752, which forms both hydrogen and hydrophobic bonds.
These three compounds have been shown to be capable of preventing PCOS with their active binding sites. A:ARG:752 is the common binding site among these three compounds (L60, L77, and L88). These three medicinal compounds are highly potential among 43 compounds. The four different types of bonds available in these interactions are the conventional hydrogen bond, the carbon-hydrogen bond, the alkyl bond, and the pi-alkyl bond. This is a critical outcome of the PCOS drug discovery with inositol, as well as crucial evidence to refer to them as highly promising drug candidates.
|
Ligand No |
Human Intestinal Absorption (+ve/-ve) |
Blood-Brain Barrier (+ve/-ve) |
Acute Oral Toxicity (kg/mol) |
Plasma protein binding |
Human oral bioavailability (+ve/-ve) |
Thyroidreceptor binding |
BSEP inhibitor |
Drug likings Rule |
Bioavailability |
Androgen receptor binding |
|
L-77 |
+0.7028 |
+0.9084 |
4.081 |
0.816 |
+0.6286 |
-0.5199 |
-0.9631 |
L,V,E,M |
0.55 |
- 0.6291 |
|
L-88 |
+0.9166 |
+0.8346 |
2.121 |
0.690 |
+0.5571 |
-0.5350 |
-0.9425 |
L,V,E,M |
0.55 |
- 0.8038 |
|
L-60 |
+0.8447 |
+0.9695 |
2.829 |
0.387 |
+0.6857 |
+0.679 6 |
-0.9565 |
L,G,V,E, M |
0.55 |
-0.6223 |
|
L-30 |
+0.9166 |
+0.8273 |
2.056 |
0.789 |
+0.5571 |
-0.6305 |
-0.9571 |
L,V,E |
0.55 |
-0.8740 |
|
L-5 |
+0.8636 |
+0.9537 |
2.371 |
0.654 |
+0.5143 |
-0.6314 |
-0.8661 |
L,V,E,M |
0.55 |
-0.7274 |
|
L-67 |
+0.9093 |
+0.9490 |
2.678 |
0.287 |
+0.6571 |
+0.583 8 |
-0.9129 |
L,V,E,M |
0.55 |
-0.6403 |
|
L-47 |
+0.8602 |
+0.9132 |
2.016 |
0.81 |
+0.5571 |
-0.4920 |
-0.9733 |
L,V |
0.55 |
-0.8653 |
|
L-33 |
+0.8899 |
+0.8326 |
1.726 |
0.793 |
+0.5143 |
-0.4900 |
-0.9767 |
L,V,E |
0.55 |
-0.9052 |
|
L-34 |
+0.8899 |
+0.8326 |
1.726 |
0.793 |
+0.5143 |
-0.4900 |
-0.9767 |
L,V,E |
0.55 |
-0.9052 |
|
L-89 |
+0.8869 |
+0.9577 |
3.176 |
0.239 |
+0.7286 |
+0.595 6 |
-0.9314 |
L,G,V,E, M |
0.55 |
-0.6366 |
|
D-1 |
+0.8263 |
+0.8032 |
2.132 |
0.654 |
+0.528 |
-0.5682 |
-0.9421 |
L,V,E |
0.55 |
-0.8052 |
|
D-2 |
+0.9943 |
+0.8862 |
1.925 |
0.724 |
+0.615 |
-0.5190 |
-0.9135 |
L,G,V,E, M |
0.55 |
-0.7056 |
|
D-3 |
+0.8103 |
+0.9012 |
2.56 |
0.680 |
+0.552 |
+0.600 1 |
-0.9056 |
L,G,V,E, M |
0.55 |
-0.9066 |
Table3: Pharmacokinetic Parameters and ADMET Property of the Best 10 Inositols.
D-1: Metformin, D-2: Spironolactone D-3: Cyproterone acetate
Admet Property Analysis
The ADMET properties analysis of 43 previous molecular docking studies’ results is in (S6). At a molecular weight above 500g/mol, molecules are absorbed more slowly in the intestines and are less likely to cross the blood-brain barrier. The permeability of a medicinal molecule is calculated by the number of hydrogen bond donors and acceptors in the molecule with a molecular weight up to 500 g/mol, H-bond donors (1-5), H-bond acceptors (1-10), and XLOGP3<4.15. No violations were found among these 43 compounds because they were already filtered from the mentioned database. Molecular complexity is dictated by the percentage of sp3 carbon atoms, which should not be lower than 0.25. Higher lipophilicity leads to decreased solubility, faster metabolism, and insufficient absorption. The polar surface area of a pharmaceutical molecule affects its bioavailability. High TPSA causes the cell membrane to be permeable. This study'sCsp3 fraction of all 43 compounds had a TPSA below 25% and low lipophilicity. The basic requirement for drug development is high biological activity with low toxicity. Each compound showed levels of toxicity III and IV. No other carcinogenic ligand (L72) will be studied further. Out of a total of 43 compounds, 42 had ADMET properties, which are suitable as prospective oral drugs. From these 42 ligands, the top 10 compounds' ADMET properties are depicted in Table 3. L77, L88, and L60 meet the ADMET properties, making them suitable for use as drugs. Molecular weight, H-bond donors, and acceptors follow the Lipinski rule. These three compounds' TPSA values are 99.38, 99.38, and 57.15Ų, and their gastrointestinal absorption is 0.55 for all three compounds. Csp3 fractions = 1.00, 1.00, and 0.67 within the normal limit. Lipinski, Veber, Egan, and Muegge rules state that L77, L88, and L60 are potent drug-like molecules.
|
Ligand No |
PubChemCID |
I=-Homo |
A=-Lumo |
Property |
|||||
|
Energy gap |
Chemical Potential |
Hardness |
Softness |
Electroneg ativity |
Electrophil icity |
||||
|
L-77 |
10858206 |
0.26572 |
-0.01581 |
0.28 |
-0.12 |
0.14 |
7.10 |
0.12 |
0.06 |
|
L-88 |
102130598 |
0.24447 |
0.02488 |
0.22 |
-0.13 |
0.11 |
9.11 |
0.13 |
0.08 |
|
L-60 |
102460862 |
0.2552 |
-0.01242 |
0.27 |
-0.12 |
0.13 |
7.47 |
0.12 |
0.06 |
Table4: Chemical Reactivity calculations of top three Ligands are assessed by Chemical Potential, Hardness, Softness, Electronegativity, Electrophilicity.
Evaluation of Chemical Reactivity by DFT
The Density Functional Theory was used to estimate the chemical stability of our target compounds by analyzing the electronic characteristics of all 42 ligands. HOMO and LUMO orbitals play a crucial role in charge transfer between these orbitals during a chemical process [39]. Chemical reactivity is reduced as the energy gap widens [48]. Table_4 shows the chemical reactivity parameters of the top three target compounds: electrophilicity (ω), chemical potential (μ), electronegativity (c), hardness (h), and softness (S). The electron gap ranges from (0.22 to 0.28) eV, where softness and hardness range from (7.10 to 9.11) eV and (0.11 to 0.14) eV. These three compounds' chemical potential and electronegativity are exactly the same (-0.12, -0.13, and -0.12) eV, where electrophilicity is0.06, 0.08, and 0.06) eV. The frontier molecular orbital of HOMO and LUMO for the compounds L77, L88, and L60 is depicted in Figure 4

Figure 4: Frontier Molecular Orbitals of Homo and Lumo for the Chemical Stability Assessment of the Compound L77, L88, andL60
After the docking, it is essential to justify and validate the docking procedure,and the molecular dynamics of docked complexes is one of the most acceptable methods. As it is a large time- consuming process, it is quite impossible to perform the MD for all docking complexes. As a result, the best-recommended inhibitors of inositol derivatives (L77 and L88) were taken to perform the MD against the protein (3RLJ), setting the range at 100 ns. Afterward, the root mean square deviation (RMSD), and root mean square fluctuation (RMSF) have accounted for checking the stability and validation of docked complex and docking procedures [49, 50]. Respectively, while the magnitude of both RMSD and RMSF is below 1.8 A°, indicating satisfactory requirements for stability and validation of docking procedures [51, 52]. As shown in Figure 5, the RMSD was illustrated against time and protein skeleton or amino acid residues in Figs (a) and (b), respectively, while the RMSD was obtained below 0.8 and 0.6 for L-77 and L-88, respectively, in terms of time dependable, and a similar trend was found for amino acid residues. Into the bargain, the RMSF with respect tothebackbone (amino acid residues up to 100 reside out of 110) of selected complexes having 100 ns was calculated, and very low fluctuations were obtained. However, it is revealed that the stability of docked complexes is very high, and it might be said that the ligand is kept in a protein pocket for 100 ns while the fluctuation is also reasonable [53].



a) RMSD with respect b) RMSD vs Amino acid c) RMSF with respect of
Time residues backbone
Figure 5: Various Pictures of Rmsd and Rmsf for Proteins are used to Check the Stability of the Docked Complex and Validate the Docking Procedure.
Ramachandran Plot from Molecular Dynamic Study
The Ramachandran plot designates the chemical stability of the protein and enzyme structures by viewing the stereochemistry and steriochemical structure, which is also supportive evidence for the validation of docking and molecular dynamic procedures [54-56]. Also, this analysis illustrates the energy minimization of Botherombin and the most favorable region. From Figure 6, the Ramachandran plotis shown for the top three drugs (L77, L88, and L60) and compared with three standard drugs (D1, D2, and D3). There are 174 amino acids (96.67%) inthefavorable region, and 6 amino acids (3.37%) intheunfavorable region, which is almost the similar trend for the D2, D3, L77, L88, and L60, meaning that there is no change after entering the ligand in protein. So that, it can be concluded that the molecular docking is valid and highly stable docked complexes were obtained after docking.
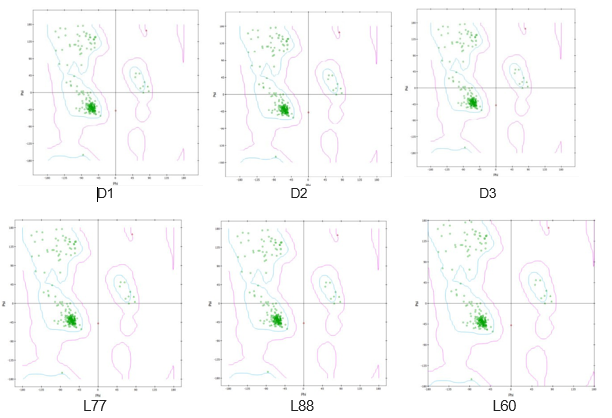
Figure 6: Picture of Ramachandran Plot for Drugs and ligands
Covariance map from Molecular Dynamic Study
The Covariance matrix indicates coupling between pairs of residues, i.e., whether they experience correlated (red), uncorrelated (white), or anti-correlated (blue) motions.
The correlation matrix is computed using the Cα Cartesian coordinates using a Covariance Analysis of Atomic Fluctuations in Molecular-Dynamics and Normal Mode Simulations [57- 59]. It is estimated that if the residue remains in the red line, they maintain the correlated and stable with valid procedure of docking as well as molecular dynamic. From the above Figure 7, about more than 95% of the residues of standard drugs (D1, D2, and D3) and ligands (L77, L88, and L60) stay in the red line. However, it can be said that the target drugs can showa similar trend compared with standard drugs from the covariance map.
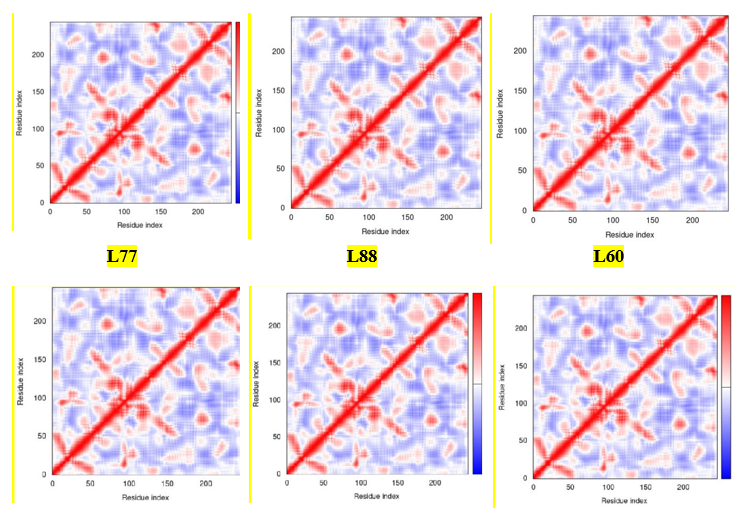
Figure 7: Covariance map from Molecular Dynamic Study
B-factor and Mobility from Molecular Dynamic Study
The expression B-factor, occasionally entitled the Debye-Waller factor, temperature factor, or atomic displacement parameter [60, 61]. It is exploited in protein crystallography to describe atomic positions from an average (mean) value (mean-square displacement) as well as the attenuation of X-ray or neutron scattering caused by thermal motion. The most significant part of it is to express that the higher flexibility results in larger displacements or lower electron density, indicating lower stability. So after molecular docking, the B-factor and mobility of the docked protein complex can be calculated, which help to say its deformation or stability at each of its residues and Normal Mode Analysis (NMA) of proteins. From Figure 8, the deformation of the docked complex has been shown, where the deformation value is almost 0.8 or less than 1.0 for all residues for L77, L88, L60, D1, D2, and D3.
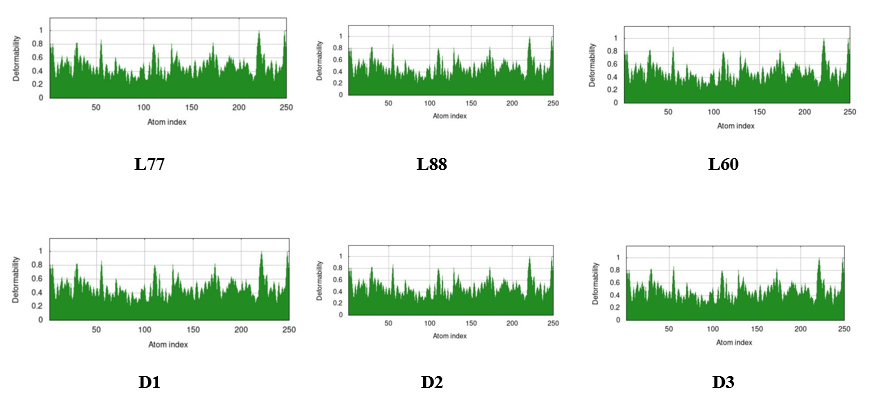
Figure 8: Deformation of B-factor and Mobility for Docked Complex
In addition, here, the experimental B-factor is taken from the corresponding Protein Data Bank (PDB) field as the reference value and calculated from NMA by multiplying the NMA mobility by (8pi^2). The rationale of the NMA is to give an averaged RMS compared to the reference PBD. From Figure 9, the B-factor for L77, L88, L60, D1, D2, and D3 is presented and compared with the experimental value of PDB. From the graph in Figure 9 for the B-factor, it can be said that all docked complexes are almost at the experimental PBD and have not obtained the large fluctuation or flexibility of atoms which mention the higher stability of them.
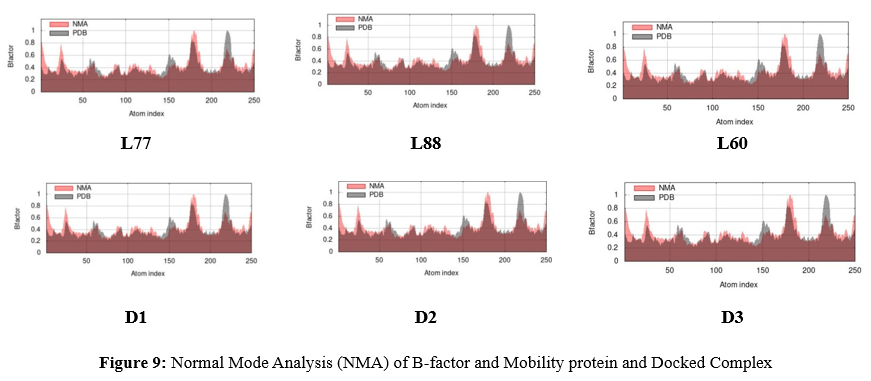
Conclusion
To prevent PCOS disorder, 6327 inositol derivatives were identified from the PubChem database stored overall110 million compounds. Out of all-up, 587 Inositols were filtered by the Lipinski Rule and pharmacokinetics. Next, 101 molecules were selected based on the ascending order of molecular weight. Ninety (90) Inositols were more responsive to testosterone through the PASS prediction study than the other two medicinal qualities. Evaluation of molecular docking study, 43 compounds were found to have a strong binding affinity (-6.0 to -7.5) against the androgen receptor protein (3RLJ) with having protein-ligand interaction analysis, bond type and number of hydrogen bonds. In addition, it wasfoundthatA:GLN:711, A:MET:745, and A:ARG:752 are the most binding sites of amino acid residues of protein, where the lowest bond distance was observed for the binding site A:GLU:681 (1.83 Å), indicating the strong potential becoming drug.
From a total of 43 substances anticipated for ADMET and carcinogenicity analysis, 42 were found to be violation-free. This group of 42 chemicals showed good biological activity to fight against PCOS illness, where chemical reactivity calculations depicted potentiality with a low energy gap and high softness value of the best three ligands. To date, no effective compounds have been approved for treatment for the androgen receptor protein to recover from PCOS illness. In this study, we are going to recommend three highly potential drug candidates (L77-1, 2-O-Cyclohexylidene-myo-inositol, L88-1-O, 4-O-Diallyl-D- myo-inositol, and L60-1-O, 2-O:3-O,4-O-Diisopropylidene-5- deoxy-L-chiro-inositol). These three compounds demonstrated high absorbability, low toxicity, and high binding affinity with the target protein -3RLJ- with proper protein-ligand interactions. In this study, however, 1,2-O-Cyclohexylidene-myo-inositol (L77) was deemed the best drug candidate in every way against PCOS, and finally, stability was confirmed by MD simulation. The MD simulation is also explained in terms of the covariance map, the Ramachandran plot, the deformation of the B-factor, the NMA of the B-factor, or mobility to check the validation of the docking procedure, the MD analysis, and the stability of the docked ligand protein complexes. From the computational and in silico study, it cannot convey the precise potential point becoming drugs or undistorted to the wet experimental value. However, further in vivo and in vitro studies are required to confirm their approval to directly use in trial for drugs.
Funding: Authors did not receive any kind of funds from any institutions even companies.
Data Availability Statement: Data will be provided by the corresponding author upon request.
Conflict of interest: The authors declare that they have no conflict of interest.
Ethical Approval: Not applicable
Informed consent: This article does not contain any studies with human participants.
References
- Ajmal, N., Khan, S. Z., & Shaikh, R. (2019). Polycystic ovary syndrome (PCOS) and genetic predisposition: A review article. European journal of obstetrics & gynecology and reproductive biology: X, 3, 100060.
- Escobar-Morreale, H. F. (2018). Polycystic ovary syndrome: definition, aetiology, diagnosis and treatment. Nature Reviews Endocrinology, 14(5), 270-284.
- Rosenfield, R. L., & Ehrmann, D.A. (2016). The pathogenesis of polycystic ovary syndrome (PCOS): the hypothesis of PCOS as functional ovarian hyperandrogenism revisited. Endocrine reviews, 37(5), 467-520.
- Haq, N., Khan, Z., Riaz, S., Nasim, A., Shahwani, R., & Tahir, M. (2017). Prevalence and knowledge of polycystic ovary syndrome (PCOS) among female science students of different public Universities of Quetta, Pakistan. Imperial Journal of Interdisciplinary Research, 35(6), 385-92.
- Kyrou, I., Karteris, E., Robbins, T., Chatha, K., Drenos, F., & Randeva, H. S. (2020). Polycystic ovary syndrome (PCOS) and COVID-19: an overlooked female patient population at potentially higher risk during the COVID-19pandemic. BMC medicine, 18, 1-10.
- Macut, D., Bjekic-Macut, J., Rahelic, D., & Doknic, M. (2017). Insulin and the polycystic ovary syndrome. Diabetes research and clinical practice, 130, 163-170.
- Witchel, S. F., Teede, H. J., & Peña, A. S. (2020). Curtailing pcos. Pediatric research, 87(2), 353-361.
- Jin, P., & Xie, Y. (2018). Treatment strategies for women with polycystic ovary syndrome. Gynecological Endocrinology, 34(4), 272-277.
- Huang, H., He, Y., Li, W., Wei, W., Li, Y., Xie, R., ... &He, W. (2016). Identification of polycystic ovary syndrome potential drug targets based on pathobiological similarity in the protein-protein interaction network. Oncotarget, 7(25), 37906.
- Walters, K. A., Allan, C. M., Jimenez, M., Lim, P. R., Davey,R. A., Zajac, J. D., ... & Handelsman, D. J. (2007). Female mice haploinsufficient for an inactivated androgen receptor (AR) exhibit age-dependent defects that resemble the AR null phenotype of dysfunctional late follicle development, ovulation, and fertility. Endocrinology, 148(8), 3674-3684.
- Rodriguez Paris, V., & Bertoldo, M. J. (2019). The mechanism of androgen actions in PCOS etiology. Medical sciences, 7(9), 89.
- Teede, H., Moran, L., Deeks, A., Chambers, D., 2008.Polycystic ovary syndrome. Aust. Dr. 25–32.
- Naz, R. K., Engle, A., & None, R. (2009). Gene knockouts that affect male fertility: novel targets for contraception. Frontiers in Bioscience (Landmark Edition), 14(10), 3994- 4007.
- Gateva, A., Unfer, V., & Kamenov, Z. (2018). The use of inositol (s) isomers in the management of polycystic ovary syndrome: a comprehensive review. Gynecological endocrinology, 34(7), 545-550.
- Genazzani, A. D. (2016). Inositol as putative integrative treatment for PCOS. Reproductive BioMedicine Online, 33(6), 770-780.
- Regidor, P. A., Schindler, A. E., Lesoine, B., & Druckman,R. (2018). Management of women with PCOS using myo- inositol and folic acid. New clinical data and review of the literature. Hormone molecular biology and clinical investigation, 34(2).
- Benelli, E., Del Ghianda, S., Di Cosmo, C., & Tonacchera,M. (2016). A combined therapy with myo-inositol and D-chiro-inositol improves endocrine parameters and insulin resistance in PCOS young overweight women. International journal of endocrinology, 2016.
- Boyle, J. A., & Teede, H. J. (2016). Refining diagnostic features in PCOS to optimize health outcomes. Nature Reviews Endocrinology, 12(11), 630-631.
- Kamenov, Z., & Gateva, A. (2020). Inositols in PCOS. Molecules, 25(23), 5566.
- Tso, L. O., Costello, M. F., Albuquerque, L. E. T., Andriolo,R. B., & Macedo, C. R. (2020). Metformin treatment before and during IVF or ICSI in women with polycystic ovary syndrome. Cochrane Database of Systematic Reviews, (12).
- Moghetti, P. (2006). Use of antiandrogens as therapy for women with polycystic ovary syndrome. Fertility and sterility, 86, S30-S31.
- Ohara, M., Yoshida-Komiya, H., Ono-Okutsu, M., Yamaguchi-Ito, A., Takahashi, T., & Fujimori, K. (2021). Metformin reduces androgen receptor and upregulates homeobox A10 expression in uterine endometrium in women with polycystic ovary syndrome. Reproductive Biology and Endocrinology, 19(1), 1-10.
- Unfer, V., Nestler, J. E., Kamenov, Z. A., Prapas, N., & Facchinetti, F. (2016). Effects of inositol (s) in women with PCOS: a systematic review of randomized controlled trials. International journal of endocrinology, 2016.
- Monastra, G., Unfer, V., Harrath, A. H., & Bizzarri, M. (2017). Combining treatment with myo-inositol and D-chiro- inositol (40: 1) is effective in restoring ovary function and metabolic balance in PCOS patients. Gynecological Endocrinology, 33(1), 1-9.
- Nordio, M., Basciani, S., & Camajani, E. (2019). The 40: 1 myo-inositol/D-chiro-inositol plasma ratio is able to restore ovulation in PCOS patients: comparison with other ratios. European Review for Medical & Pharmacological Sciences, 23(12).
- Facchinetti, F., Appetecchia, M., Aragona, C., Bevilacqua, A., Bezerra Espinola, M. S., Bizzarri, M., ... & Unfer, V. (2020). Experts’ opinion on inositols in treating polycystic ovary syndrome and non-insulin dependent diabetes mellitus: A further help for human reproduction and beyond. Expert opinion on drug metabolism & toxicology, 16(3), 255-274.
- Facchinetti, F., Unfer, V., Dewailly, D., Kamenov, Z. A., Diamanti-Kandarakis, E., Laganà, A. S., ... & Soulage,C. O. (2020). Inositols in polycystic ovary syndrome: An overview on the advances. Trends in Endocrinology & Metabolism, 31(6), 435-447.
- Wojciechowska, A., Osowski, A., Józwik, M., Górecki, R., Rynkiewicz, A., & Wojtkiewicz, J. (2019). Inositols’ importance in the improvement of the endocrine–metabolic profile in PCOS. International journal of molecular sciences, 20(22), 5787.
- Larvol, B. L., & Wilkerson, L. J. (1998). In silico drug discovery: tools for bridging the NCE gap. Nature Biotechnology, 16(1), 33-34.
- Opo, F. A. D. M., Rahman, M. M., Ahammad, F., Ahmed, I., Bhuiyan, M. A., & Asiri, A. M. (2021). Structure based pharmacophore modeling, virtual screening, molecular docking and ADMET approaches for identification of natural anti-cancer agents targeting XIAP protein. Scientific reports, 11(1), 4049.
- Trisnawati, Y., Suminto, Sudaryono, A., 2019. Fo r P ee r R ev iew Fo r P ee r R. Neurogastroenterol. Motil.
- Tahir ul Qamar, M., Maryam, A., Muneer, I., Xing, F., Ashfaq, U. A., Khan, F. A., ... & Siddiqi, A. R. (2019). Computational screening of medicinal plant phytochemicals to discover potent pan-serotype inhibitors against dengue virus. Scientific reports, 9(1), 1433.
- Duke III, C. B., Jones, A., Bohl, C. E., Dalton, J. T., & Miller,D. D. (2011). Unexpected binding orientation of bulky-B- ring anti-androgens and implications for future drug targets. Journal of medicinal chemistry, 54(11), 3973-3976.
- Yuan, S., Chan, H. S., & Hu, Z. (2017). Using PyMOL as a platform for computational drug design. Wiley Interdisciplinary Reviews: Computational Molecular Science, 7(2), e1298.
- Islama, M., Kumerb, A., Sarkera, N., interactions, S.P.-, 2019, undefined, 2019. The prediction and theoretical study for chemical reactivity, thermophysical and biological activity of morpholinium nitrate and nitrite ionic liquid crystals: A DFT study. Adv. J. Chem. A 2, 316–326. https:// doi.org/10.33945/sami/ajca.2019.4.5
- Kawsar, S., & Kumer, A. (2021). Computational investigation of methyl α-D-glucopyranoside derivatives as inhibitor against bacteria, fungi and COVID-19 (SARS-2). Journal of the Chilean Chemical Society, 66(2), 5206-5214.
- Kumer, A., Chakma, U., Rana, M. M., Chandro, A.,Akash, S., Elseehy, M. M., ... & El-Shehawi, A. M. (2022). Investigation of the new inhibitors by sulfadiazine and modified derivatives of α-d-glucopyranoside for white spot syndrome virus disease of shrimp by in silico: quantum calculations, molecular docking, ADMET and molecular dynamics study. Molecules, 27(12), 3694.
- Kumer, A., Sarker, M. N., & Paul, S. (2019). The thermo physical, HOMO, LUMO, Vibrational spectroscopy and QSAR study of morphonium formate and acetate Ionic Liquid Salts using computational method. Turkish Computational and Theoretical Chemistry, 3(2), 59-68.
- Tripathy, S., Sahu, S. K., Azam, M. A., & Jupudi, S. (2019). Computer-aided identification of lead compounds as Staphylococcal epidermidis FtsZ inhibitors using molecular docking, virtual screening, DFT analysis, and molecular dynamic simulation. Journal of Molecular Modeling, 25, 1-13.
- Genc, Z. K., Tekin, S., Sandal, S., Sekerci, M., & Genc,M. (2015). Synthesis and DFT studies of structural and some spectral parameters of nickel (II) complex with 2-(2-hydroxybenzoyl)-N-(1-adamantyl) hydrazine carbothioamide. Research on Chemical Intermediates, 41, 4477-4488.
- Najar, A. M., Taynaz, M. A., Elmarzugi, N. A., Atia, A. E., & Moftah, S. A. M. (2020). COVID-19 Drug Design Based on the Active Core of Well-known Anti-malarial: A computational Approaches J Theor Comput Sci. 6: 266.
- Ahammad, F., Alam, R., Mahmud, R., Akhter, S.,Talukder, E. K., Tonmoy, A. M., ... & Qadri, I. (2021). Pharmacoinformatics and molecular dynamics simulation- based phytochemical screening of neem plant (Azadiractha indica) against human cancer by targeting MCM7 protein. Briefings in Bioinformatics, 22(5), bbab098.
- Rahman, M. M., Biswas, S., Islam, K. J., Paul, A. S., Mahato, S. K., Ali, M. A., & Halim, M. A. (2021). Antiviral phytochemicals as potent inhibitors against NS3 protease of dengue virus. Computers in Biology and Medicine, 134, 104492.
- Rakib, A., Nain, Z., Sami, S. A., Mahmud, S., Islam, A., Ahmed, S., ... & Simal-Gandara, J. (2021). A molecular modelling approach for identifying antiviral selenium- containing heterocyclic compounds that inhibit the main protease of SARS-CoV-2: An in silico investigation. Briefings in bioinformatics, 22(2), 1476-1498.
- Nath, A., Kumer, A., Zaben, F., & Khan, M. W. (2021). Investigating the binding affinity, molecular dynamics, and ADMET properties of 2, 3-dihydrobenzofuran derivatives as an inhibitor of fungi, bacteria, and virus protein. Beni- Suef University Journal of Basic and Applied Sciences, 10(1), 1-13.
- Agahi, F., Juan, C., Font, G., & Juan-García, A. (2020). In silico methods for metabolomic and toxicity prediction of zearalenone, α-zearalenone and β-zearalenone. Food and Chemical Toxicology, 146, 111818.
- Salmaso, V., & Moro, S. (2018). Bridging molecular docking to molecular dynamics in exploring ligand- protein recognition process: An overview. Frontiers in pharmacology, 9, 923.
- Uzzaman, M., & Hoque, M. J. (2018). Physiochemical, molecular docking, and pharmacokinetic studies of Naproxen and its modified derivatives based on DFT. Int. J. Sci. Res. Manag, 6, 2018-2025.
- Hashem, H. E., Mohamed, E. A., Farag, A. A., Negm, N. A., & Azmy, E. A. (2021). New heterocyclic Schiff base-metal complex: Synthesis, characterization, density functional theory study, and antimicrobial evaluation. Applied Organometallic Chemistry, 35(9), e6322.
- Nath, A., Kumer, A., Zaben, F., & Khan, M. W. (2021). Investigating the binding affinity, molecular dynamics, and ADMET properties of 2, 3-dihydrobenzofuran derivatives as an inhibitor of fungi, bacteria, and virus protein. Beni- Suef University Journal of Basic and Applied Sciences, 10(1), 1-13.
- Kumer, A., Chakma, U., Matin, M. M., Akash, S., Chando, A., & Howlader, D. (2021). The computational screening of inhibitor for black fungus and white fungus by D-glucofuranose derivatives using in silico and SAR study. Organic Communications, 14(4).
- Rahman, M. A., Matin, M. M., Kumer, A., Chakma, U., &Rahman, M. R. (2022). Modified D-glucofuranoses as new black fungus protease inhibitors: Computational screening, docking, dynamics, and QSAR study. Physical Chemistry Research, 10(2), 195-209.
- Kumer, A., Chakma, U., & Kawsar, S. M. A. (2021). The inhibitory effect of somenaturalbioactive D-glucopyranoside derivatives against SARS-CoV-2 main protease (Mpro) and spike protease (Spro). ASM Sci. J, 16, 1-18.
- Hooft, R. W., Sander, C., & Vriend, G. (1997). Objectively judging the quality of a protein structure from a Ramachandran plot. Bioinformatics, 13(4), 425-430.
- Rani, M., Nath, A., & Kumer, A. (2023). In-silico investigations on the anticancer activity of selected 2-aryloxazoline derivatives against breast cancer. Journal of Biomolecular Structure and Dynamics, 41(17), 8392-8401.
- Sheik, S. S., Sundararajan, P., Hussain, A. S. Z., & Sekar,K. (2002). Ramachandran plot on the web. Bioinformatics,18(11), 1548-1549.
- Agarwal, P., Tipaldi, G. D., Spinello, L., Stachniss, C., & Burgard, W. (2013, May). Robust map optimization using dynamic covariance scaling. In 2013 IEEE International Conference on Robotics and Automation (pp. 62-69). Ieee.
- Frasinski, L. J. (2016). Covariance mapping techniques. Journal of Physics B: Atomic, Molecular and Optical Physics, 49(15), 152004.
- Ichiye, T., & Karplus, M. (1991). Collective motions in proteins: a covariance analysis of atomic fluctuations in molecular dynamics and normal mode simulations. Proteins: Structure, Function, and Bioinformatics, 11(3), 205-217.
- Barthels, F., Schirmeister, T., & Kersten, C. (2021). BANΔIT: B’-factor analysis for drug design and structural biology. Molecular Informatics, 40(1), 2000144.
- Yuan, Z., Bailey, T. L., & Teasdale, R. D. (2005). Prediction of protein B-factor profiles. Proteins: Structure, Function, and Bioinformatics, 58(4), 905-912.