
Journal of Clinical & Experimental Immunology(JCEI)
ISSN: 2475-6296 | DOI: 10.33140/JCEI
Impact Factor: 1.9

Review Article - (2025) Volume 10, Issue 2
HLA-1-opathies and peptides
Received Date: Oct 31, 2025 / Accepted Date: Nov 26, 2025 / Published Date: Dec 12, 2025
Copyright: ©©2025 James R Archer. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Citation: Archer, J. R. (2025). HLA-1-opathies and peptides. J Clin Exp Immunol, 10(2), 01-06
Abstract
It is almost 50 years since it was realised that specific HLA molecules are associated with numerous chronic inflammatory diseases [1]. The HLA-1-opathies were subsequently defined as a subgroup of these, characterized genetically by three specific genes - a class I HLA which controls the particular disease, ERAP1, coding an endopeptidase trimming the peptides included in the class I heterotrimer and IL23R, coding part of the IL23 receptor, which affects the strength and nature of the inflammatory response [2]. Originally, the most obvious association was of the class I molecule HLA B27 with the arthritic disease ankylosing spondylitis (AS), which is now included in axial spondyloarthropathy (axSpa) [3,4]. As it was assumed that HLA molecules were involved in protection from infection, extensive efforts were made to identify microbial agents causing these diseases. Currently, however, although many associations are still being found, no foreign agent has been identified as a plausible explanation for the pathology. This paper offers an alternative hypothesis, questioning the assumption that cells invariably produce sufficient peptides to complete HLA class 1 heterotrimers and emphasising the role of self proteins, together with explanations for why the microbial data have caused so much confusion. It suggests new targets for the development of therapies.
Graphical Abstarct
Alternative variations of HLA Class 1 molecules leading to inflammation
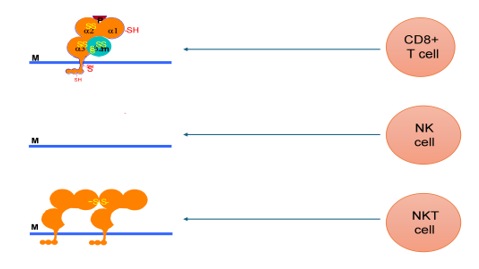
Top: HLA B27 molecules presenting viral peptide
Middle: missing HLA B27 molecule
Bottom: modified B27 molecule
M cell membrane
Main
HLA class I molecules have two functions. First, they present peptides from internal pathogens to the cell surface for recognition by CD8+ cytotoxic T cells, causing these infected cells to be killed. Secondly, they protect cells from killing by NK cells by presenting self-peptides produced by healthy cells [5]. A variant of this process is that some cells carrying killer-cell Ig-like receptors (NKT cells) recognise misfolded class I molecules [6]. These second activities tend to negate the common viral strategy of avoiding T cell killing by interfering with the maturation or expression of class I molecules [7]. Most attempts to explain class I associated disease have assumed an association with the first function. This paper suggests that we should consider the effects of class I processing of self-peptides in normal cells on NK and NKT cells. It depends on the hypothesis that disease is caused either by minimal availability of suitable peptides or late processing of peptides by endoplasmic reticulum peptidases, producing peptides which fit poorly in class I heterotrimers and making them unstable, so that they are either missing from the cell surface or arrive as free heavy chains or misfolded molecules. Cell surfaces either lacking suitably-folded class 1 heterotrimers or presenting misfolded molecules at the surface will trigger an inflammatory NK or NKT cell response. The paper also offers explanations for previous confusion involving foreign peptides and suggests that evidence of a role for microbial cross-reaction has been misinterpreted.
The HLA-1-opathies are characterised by the activities of three genes – IL23R, ERAP1 and a class I HLA. IL23R genes ensure a strong IL23 / IL17 inflammatory response and ERAP1 codes for endoplasmic reticulum peptidases which trim the peptides of class I molecules [2]. Specific class I genes code for individual diseases, notably B*27, particularly associated with axial spondyloarthropathy (axSpa), C*0602 with psoriasis (Ps), B*51 with Behçet's disease and A*2902 with Birdshot Retinopathy (BR) [3,8-10]. The evidence in this paper focuses largely on B*27, whose biochemistry has been extensively studied.
Several animal models have been developed by inserting a human class I gene. A B27 mouse model suggested that arthritis could be caused by the B*27 heavy chain alone [11]. An A29 mouse developed retinal inflammation described as resembling several CD4+ T cell driven retinal conditions [12,13]. The B27 rat has been extensively investigated and produced unexpected results which have tended to be ignored ‘because it’s a rat’. Notably, it was early demonstrated that although CD4+ T cells are required for rats to develop arthritis there is no need for the CD8+ cytotoxic T cells which recognise virus-peptide charged class I molecules [14,15].
This fits poorly with the notion that AS is associated with specific cytotoxic T cell immunity. Secondly, insertion into the B27 rat genome of a gene for a viral peptide to be expressed in quantity in the endoplasmic reticulum (ER) prevented B27-associated arthritis [16]. This showed that peptides, or their absence, were important in disease development and peptides not associated with infection could modify the disease. Finally, B27 rats, like human patients, develop a gut inflammation as well as arthritis. It was shown that the presence of extra human β2-microglobulin in the rat enhanced the arthritis [17].
This suggests that the gut and bone inflammation are related conditions but do not have identical mechanisms and, although they may confuse interpretations, one does not lead to the other. Here it is proposed that gut inflammation is caused by reactions within the endoplasmic reticulum (ER) but the B27 molecules associated with arthritis requiring extra β2-microglobulin in the rat are more mature and have progressed further through the cell.

Figure1: Peptide-Binding Grove of HLA B27
Red Heavy chain – β2 micro globulin stabilisation
Green F pocket sites affecting susceptibility
Yellow Cysteines
A fully-folded class I molecule consists of a heavy chain containing a groove formed by its a1 and a2 domains holding a peptide of 8-12 amino acids and stabilised by the peptide and β2-microglobulin to form a heterotrimer [18]. Regions within the groove which associate with individual amino acids are labelled pockets. Position 97 of the class I heavy chain helps to stabilise its interaction with β2-microglobulin [19]. HLA-1-opathies occur in those variants at this position in which this is least effective [20]. HLA*B27 includes two subtypes, B*2706 and B*2709, which are not associated AS, and differ respectively from the arthritis- associated B2704 and B2705 subtypes by single amino acids in the F pocket of the peptide-binding groove [21]. They fold more rapidly as they progress through the endoplasmic reticulum and peptide loading complex [22].
The B pocket includes a reactive free cysteine at position 67, present in all B*27 subtypes, able to form homodimers through disulfide bonds on the cell surface which react with NKT cells to induce production of inflammatory molecules typical of HLA- 1-opathies [23,24]. Inappropriate dimers can also be formed by reactions of the cysteines in the α2 domain (C101 and C164) both with C67 and with each other [25]. These can be expressed either internally or externally [26]. Inappropriate dimers occur in those subtypes which tend to fold slowly [27].
Folding rates will depend on the availability of suitable peptides and the ultimate stability of the molecule is likely to depend on the fit and concentration of individual peptides in the final stages of synthesis. The specific self-peptides are determined by the cells which produce them; but currently nearly all our information on HLA synthesis and expression is derived from cultured fibroblasts, leucocyte cell lines, HeLa cells or freshly isolated blood cells [28]. This hypothesis suggests that in the specific case of HLA-B*27, protective peptide availability is less in gut and bone cells. In the gut this decreases the availability of well-fitting peptides in the ER.
It makes folding more difficult, adds to the likelihood of production of dimers and permits misfolding to the point that the molecules trigger unfolded protein (UPR) or ER overload (EOR) responses, leading to gut inflammation and leakage [26]. It is suggested that, in the rat model, large quantities of β2-microglobulin aid folding in the gut but fail to protect bone cells from poorly or over- trimmed specific peptides arriving at the cell surface. This process will cause either NK or NKT responses. These conclusions are consistent with a functional genomic approach which concluded that the principal cells involved in axial spondyloarthropathy are NK cells and the observation that B27 homodimers stimulate an NKT cell response [24,29].
It is key to this hypothesis that proteins which cause significant loss of HLA heterotrimers are specifically produced in significantly altered amounts in cells within the tissues where inflammation occurs. Indications of the identity of these cells is the suspicion that AS is triggered in stressed joints which are affected by constant repair and remodelling [30]. Likewise, in psoriasis, stimulation of the skin by a pinprick can cause a sterile neutrophil response (Köbner response) and the pathergy response is a similar reaction occurring in Behçet's disease [31,32].
Currently, no mechanism seems to have been offered for differences in rates of folding of different B27 subtypes. One possibility is that most B27 subtypes have difficulty in acquiring suitable peptides. If a subtype such as B2706 binds more easily with a peptide derived from a house-keeping protein it will fold faster. The presence of that protein in gut cells will protect from the UPR or EOR, whereas in bone cells it will allow production of sufficient heterotrimers to protect them from NK or NKT cells.
A recurring theme of research into spondyloarthropathies has been the attempt to relate them to infections and a role for bacteria in B27-associated arthritis impossible to ignore. Germ-free B27 rats do not develop arthritis until they have developed a commensal bacterial population [33]. It is suggested that this occurs because of a requirement for commensal bacteria for normal immune cell development rather than a specific infection [34]. Reactive arthritis, which is frequently associated with B*27, can be triggered by Chlamydia and number of unrelated agents, including Enterobacteriaceae such as Shigella and Pasteurella. The arthritis is unaffected by treatments expected to cure the infection and persists for long after the infection has disappeared [35,36]. Substantial attempts to show large numbers of cross-reactive cytotoxic T cell responses involving Chlamydia also failed [37].
Antibodies, including a monoclonal antibody reacting with some B27 molecules and Klebsiella, were shown to react with outer membrane proteins of Gram-negative bacteria, but could never be shown to be associated with AS [38]. None of these observations is consistent with a role for an adaptive immune response in AS. A further problem in interpreting results is the tendency of immunologically-activated cells to relocate to other sites of chronic inflammation, so cells activated by bacteria at a leaking gut are likely to confuse interpretations by seeking out an inflamed joint [39,40].
Recent research has stressed the role the gut biota in promoting or permitting arthritis [41]. These results are complex and sometimes contradictory [42]. It seems likely that there is an interplay between inflammatory cells and bacteria altering the chemistry of the alimentary tract which may, in turn, alter the activity and locations of inflammatory cells. None of the bacteria currently under study was associated with the original reactive arthritis observations. However, it may be that infections triggering reactive arthritis alter gut populations in such a way that they render patients more susceptible to inflammation.
The recent apparent cure of a patient with AS by treatment with antibodies to a CD8+ T cell receptor motif requires comment [43]. The treatment was based on the identification of a group of CD8+ T cell receptors whose proportions were enhanced in B27- positive patients with either AS or acute anterior uveitis [44]. These proportions were further increased in the relevant inflamed tissues. CD8+ T cell receptors are relatively non-specific and a number of their predicted targets could be expressed on a class 1 deficient cell line. Those peptides included some derived from human genes, none predicted to be unusually well expressed in the specific inflamed tissues, as well as several derived from gut- associated Enterobacteriaceae. So far as they were measured, the antibacterial reactions of inflamed tissue T cells seem to have been slightly higher against a bacterial peptide than against human self-peptides, so they may have been derived from activated cells in the inflamed gut migrating to a second site of inflammation. One possibility is that the extra concentration of cytotoxic cells in inflamed tissues derived from anti-bacterial T cells was sufficient to cause further inflammation in an already-stressed tissue.
In summary, it is hypothesised that HLA-1-opathies are caused by deficiencies in appropriate peptides or overloading of nascent specific class I molecules by poorly fitting peptides produced in large amounts in specific tissues. Loss or modification of class I triggers an antiviral NK or NKT cell response. Disease activity may be further modified by the presence of specific T cells attracted to the site by the initial inflammatory response. The hypothesis explains tissue specificity, the difference between arthritogenic and non-arthritogenic HLA-B27 variants, associations with repair processes and some anomalous B27 rat results.
The hypothesis emphasises the importance of self, rather than foreign, peptides. There is value in identifying any causing heterotrimer instability. They should be recognisable by their inclusion of peptides relevant to the specific HLA specificities involved, their presence in significant quantities in specific tissues and by their changes in their concentration during repair processes [45,46]. Currently, research into treatment of HLA-1-opathies almost exclusively involves use of anti-inflammatory molecules [47]. Alternatives would be to look for ways of altering presence of arthritogenic self-peptides or increasing the availability of safe self-peptides.
References
- Dausset, J., & Svejgaard, A. (1976). HLA and disease:predisposition to disease and clinical implications. Inserm.
- McGonagle, D., Aydin, S. Z., Gül, A., Mahr, A., & Direskeneli, H. (2015). 'MHC-I-opathy'—unified concept for spondyloarthritis and Behçet disease. Nature Reviews Rheumatology, 11(12), 731-740.
- Brewerton, D. A., Hart, F. D., Nicholls, A., Caffrey, M., James,D. C. O., & Sturrock, R. D. (1973). Ankylosing spondylitis and HL-A 27. The Lancet, 301(7809), 904-907.
- Rudwaleit, M., & Taylor, W. J. (2010). Classification criteria for psoriatic arthritis and ankylosing spondylitis/ axial spondyloarthritis. Best Practice & Research Clinical Rheumatology, 24(5), 589-604.
- Djaoud, Z., & Parham, P. (2020). HLAs, TCRs, and KIRs, a triumvirate of human cell-mediated immunity. Annual Review of Biochemistry, 89(1), 717-739.
- Bowness, P., Ridley, A., Shaw, J., Chan, A. T., Wong- Baeza, I., Fleming, M., ... & Kollnberger, S. (2011). Th17 cells expressing KIR3DL2+ and responsive to HLA-B27homodimers are increased in ankylosing spondylitis. The Journal of Immunology, 186(4), 2672-2680.
- Tortorella, D., Gewurz, B. E., Furman, M. H., Schust, D. J., & Ploegh, H. L. (2000). Viral subversion of the immune system. Annual review of immunology, 18(1), 861-926.
- Owczarek, W. (2022). The role of HLA-Cw6 in psoriasis and psoriatic arthritis. Reumatologia/Rheumatology, 60(5), 303-305.
- Al-Obeidi, A. F., & Nowatzky, J. (2023). Immunopathogenesis of Behçet's disease. Clinical Immunology, 253, 109661.
- LeHoang, P., Ozdemir, N., Benhamou, A., Tabary, T., Edelson, C., Betuel, H., ... & Cohen, J. H. (1992). HLA-A29. 2 subtypes associated with birdshot retinochoroidopathy. American journal of ophthalmology, 113(1), 33-35.
- Khare, S. D., Bull, M. J., Hanson, J., Luthra, H. S., & David,C. S. (1998). Spontaneous inflammatory disease in HLA-B27 transgenic mice is independent of MHC class II molecules: a direct role for B27 heavy chains and not B27-derived peptides. The Journal of Immunology, 160(1), 101-106.
- Szpak, Y., Vieville, J. C., Tabary, T., Naud, M. C., Chopin, M., Edelson, C., ... & Pla, M. (2001). Spontaneous retinopathy in HLA-A29 transgenic mice. Proceedings of the National Academy of Sciences, 98(5), 2572-2576.
- De Kozak, Y., Camelo, S., & Pla, M. (2008). Pathological aspects of spontaneous uveitis and retinopathy in HLA-A29 transgenic mice and in animal models of retinal autoimmunity: relevance to human pathologies. Ophthalmic Research, 40(3- 4), 175-180.
- Breban, M., Fernández-Sueiro, J. L., Richardson, J. A., Hadavand, R. R., Maika, S. D., Hammer, R. E., & Taurog, J.D. (1996). T cells, but not thymic exposure to HLA-B27, are required for the inflammatory disease of HLA-B27 transgenic rats. The Journal of Immunology, 156(2), 794-803.
- May, E., Dorris, M. L., Satumtira, N., Iqbal, I., Rehman, M. I., Lightfoot, E., & Taurog, J. D. (2003). CD8αβ T cells are not essential to the pathogenesis of arthritis or colitis in HLA-B27 transgenic rats. The Journal of Immunology, 170(2), 1099- 1105.
- Zhou, M., Sayad, A., Simmons, W. A., Jones, R. C., Maika, S. D., Satumtira, N., ... & Taurog, J. D. (1998). The specificity of peptides bound to human histocompatibility leukocyte antigen (HLA)-B27 influences the prevalence of arthritis in HLA-B27 transgenic rats. The Journal of experimental medicine, 188(5), 877-886.
- Tran, T. M., Dorris, M. L., Satumtira, N., Richardson, J. A., Hammer, R. E., Shang, J., & Taurog, J. D. (2006). Additional human β2â?microglobulin curbs HLA–B27 misfolding and promotes arthritis and spondylitis without colitis in male HLA–B27–transgenic rats. Arthritis & Rheumatism, 54(4), 1317-1327.
- Bjorkman, P. J., Saper, M. A., Samraoui, B., Bennett, W. S., Strominger, J. T., & Wiley, D. C. (1987). Structure of the human class I histocompatibility antigen, HLA-A2. Nature, 329(6139), 506-512.
- Chen, L., Shi, H., Yuan, J., & Bowness, P. (2017). Position 97 of HLA-B, a residue implicated in pathogenesis of ankylosingspondylitis, plays a key role in cell surface free heavy chain expression. Annals of the Rheumatic Diseases, 76(3), 593- 601.
- Cortes, A., Pulit, S. L., Leo, P. J., Pointon, J. J., Robinson,P. C., Weisman, M. H., ... & Brown, M. A. (2015). Major histocompatibility complex associations of ankylosing spondylitis are complex and involve further epistasis with ERAP1. Nat Commun 6: 7146. doi. org/10.1038/ncomm s814, 6.
- Khan, M. A. (2000). HLA-B27 polymorphism and association with disease. The Journal of rheumatology, 27(5), 1110-1114.
- Guiliano, D. B., North, H., Panayoitou, E., Campbell, E. C., McHugh, K., Cooke, F. G., ... & Antoniou, A. N. (2017). Polymorphisms in the F Pocket of HLA–B27 Subtypes Strongly Affect Assembly, Chaperone Interactions, and Heavyâ?Chain Misfolding. Arthritis & rheumatology, 69(3), 610-621.
- Kollnberger, S., & Bowness, P. (2009). The role of B27 heavy chain dimer immune receptor interactions in spondyloarthritis. Molecular Mechanisms of Spondyloarthropathies, 277-285.
- Chan, A. T., Kollnberger, S. D., Wedderburn, L. R., & Bowness, P. (2005). Expansion and enhanced survival of natural killer cells expressing the killer immunoglobulinâ? like receptor KIR3DL2 in spondylarthritis. Arthritis & Rheumatism, 52(11), 3586-3595.
- Antoniou, A. N., Ford, S., Taurog, J. D., Butcher, G. W., & Powis, S. J. (2004). Formation of HLA-B27 homodimers and their relationship to assembly kinetics. Journal of Biological Chemistry, 279(10), 8895-8902.
- Antoniou, A. N., Lenart, I., & Guiliano, D. B. (2011). Pathogenicity of misfolded and dimeric HLAâ?B27 molecules. International journal of rheumatology, 2011(1), 486856.
- Lenart, I., Truong, L. H., Nguyen, D. D., RasiukienÄ?, O., Tsao, E., Armstrong, J., ... & Antoniou, A. N. (2022). Intrinsic folding properties of the hla-B27 heavy chain revealed by single chain trimer versions of peptide-loaded class I major histocompatibility complex molecules. Frontiers in immunology, 13, 902135.
- Sesma, L., Alvarez, I., Marcilla, M., Paradela, A., & de Castro,J. A. L. (2003). Species-specific differences in proteasomal processing and tapasin-mediated loading influence peptide presentation by HLA-B27 in murine cells. Journal of Biological Chemistry, 278(47), 46461-46472.
- Chiñas, M., Fernandez-Salinas, D., Aguiar, V. R., Nieto- Caballero, V. E., Lefton, M., Nigrovic, P. A., ... & Gutierrez- Arcelus, M. (2025). Functional genomics implicates natural killer cells in the pathogenesis of ankylosing spondylitis. Human Genetics and Genomics Advances, 6(1).
- Benjamin, M., Toumi, H., Suzuki, D., Redman, S., Emery, P., & McGonagle, D. (2007). Microdamage and altered vascularity at the enthesis–bone interface provides an anatomic explanation for bone involvement in the HLA–B27– associated spondylarthritides and allied disorders. Arthritis & Rheumatism, 56(1), 224-233.
- Langevitz, P., Buskila, D., & Gladman, D. D. (1990). Psoriatic arthritis precipitated by physical trauma. The Journal of rheumatology, 17(5), 695-697.
- Dinc, A., Karaayvaz, M., Caliskaner, A. Z., Pay, S., Erdem, H., & Turan, M. (2000). Dermographism and atopy in patients with Behçet's disease. Journal of investigational allergology & clinical immunology, 10(6), 368-371.
- Taurog, J. D., Richardson, J. A., Croft, J. T., Simmons, W. A., Zhou, M., Fernández-Sueiro, J. L., ... & Hammer, R. E. (1994). The germfree state prevents development of gut and joint inflammatory disease in HLA-B27 transgenic rats. The Journal of experimental medicine, 180(6), 2359-2364.
- Sonnenberg, G. F., & Artis, D. (2012). Innate lymphoid cell interactions with microbiota: implications for intestinal health and disease. Immunity, 37(4), 601-610.
- Gaston, J. S. (2004). Reactive arthritis and enteropathic arthropathy. 2013:901–10.
- Gaston, J.S.H. (2013). Reactive arthritis and enteropathy arthropathy. Rheumatology, Fourth Edition. Oxford: Oxford University Press,:901–10.
- Appel, H., Kuon, W., Kuhne, M., Wu, P., Kuhlmann, S., Kollnberger, S., ... & Sieper, J. (2004). Use of HLA-B27 tetramers to identify low-frequency antigen-specific T cells in Chlamydia-triggered reactive arthritis. Arthritis Res Ther, 6(6), R521.
- Yu, D. T., Choo, S. Y., & Schaack, T. (1989). Molecular mimicry in HLA-B27-related arthritis. Annals of internal medicine, 111(7), 581-591.
- Taylor-Robinson, D., & Keat, A. (2001). How can a causal role for small bacteria in chronic inflammatory arthritides be established or refuted?. Annals of the rheumatic diseases, 60(3), 177-185.
- Scotet, E., Peyrat, M. A., Saulquin, X., Retiere, C., Couedel, C., Davodeau, F., ... & Bonneville, M. (1999). Frequent enrichment for CD8 T cells reactive against common herpes viruses in chronic inflammatory lesions: towards a reassessment of the physiopathological significance of T cell clonal expansions found in autoimmune inflammatory processes. European journal of immunology, 29(3), 973-985.
- Simone, D., Al Mossawi, M. H., & Bowness, P. (2018). Progress in our understanding of the pathogenesis of ankylosing spondylitis. Rheumatology, 57(suppl_6), vi4-vi9.
- Stoll, M. L., Sawhney, H., Wells, P. M., Sternes, P. R.,Reveille, J. D., Morrow, C. D., ... & Gensler, L. S. (2023). The faecal microbiota is distinct in HLA-B27+ ankylosing spondylitis patients versus HLA-B27+ healthy controls. Clin Exp Rheumatol, 41(5), 1096-1104.
- Britanova, O. V., Lupyr, K. R., Staroverov, D. B., Shagina, I. A., Aleksandrov, A. A., Ustyugov, Y. Y., ... & Chudakov, D. M. (2023). Targeted depletion of TRBV9+ T cells as immunotherapy in a patient with ankylosing spondylitis. Nature medicine, 29(11), 2731-2736.
- Yang, X., Garner, L. I., Zvyagin, I. V., Paley, M. A., Komech,E. A., Jude, K. M., ... & Garcia, K. C. (2022). Autoimmunity- associated T cell receptors recognize HLA-B* 27-bound peptides. Nature, 612(7941), 771-777.