
International Journal of Clinical and Medical Education Research(IJCMER)
ISSN: 2832-7705 | DOI: 10.33140/IJCMER
Impact Factor: 0.93


Research Article - (2025) Volume 4, Issue 2
Exploring the Binding Profiles of Natural Compounds and BTK Inhibitors: A Docking and Electrostatic Study
Received Date: Mar 05, 2025 / Accepted Date: Apr 03, 2025 / Published Date: Apr 07, 2025
Copyright: ©Ã?©2025 Alessandro Careglio. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Citation: Careglio, A. (2025). Exploring the Binding Profiles of Natural Compounds and BTK Inhibitors: A Docking and Electrostatic Study. Int J Clin Med Edu Res, 4(2), 01-09.
Abstract
Bruton's tyrosine kinase (BTK) is a cytoplasmic, non-receptor protein tyrosine kinase crucial in B-cell signaling and a key target for treating hematological malignancies and inflammatory diseases. This study investigates the interactions between natural compounds and BTK inhibitors using computational docking and electrostatic complementarity analyses. We performed in silico docking studies with Flare Cresset, comparing known BTK inhibitors with compounds from various natural sources. Results indicate several natural compounds exhibit significant binding scores to BTK, suggesting potential as BTK modulators. Notably, Hypericum compounds showed high affinity for serum albumin, potentially leading to drug displacement. Additionally, quinone and anthraquinone compounds from species like St. John's wort, Rumex, and aloe demonstrated promising interactions with BTK. This research provides insights into natural compounds as potential BTK modulators and highlights the importance of considering drug-herb interactions. It also underscores the need for further experimental studies to validate these findings and explore their therapeutic potential.
Keywords
Bruton's Tyrosine Kinase (BTK) Inhibitors, Herbal Compounds, Drug-Herb Interactions, Computational Docking, Electrostatic Complementarity, Natural Product Interactions.
Graphical Abstract
Introduction
Bruton's tyrosine kinase (BTK) is a cytoplasmic, non-receptor protein tyrosine kinase belonging to the Tec kinase family. First discovered in X-linked agammaglobulinemia, BTK is expressed in various hematopoietic cells, excluding plasma cells, natural killer cells, and T lymphocytes. While initially recognized for its critical role in B-cell adaptive immunity, recent studies have highlighted its crucial role in innate immunity [1].
BTK regulates signaling downstream of the B-cell receptor (BCR), Fc receptors (FcRs), and Toll-like receptors, making it an attractive drug target. Inhibition of BTK activity has been shown to reduce multi-organ injury/dysfunction (cardiac, renal, hepatic) caused by trauma, and BTK inhibitors have been investigated for mitigating the cytokine storm and lung injury associated with severe acute respiratory syndrome caused by coronavirus 2 (SARS-CoV-2) [2].
BTK plays a central role in the signal transduction of the B-cell antigen receptor and other cell surface receptors, both in normal and malignant B lymphocytes. B-cell antigen receptor signaling is activated in secondary lymphatic organs and drives the proliferation of malignant B cells, including chronic lymphocytic leukemia (CLL) cells. Consequently, BTK has emerged as a promising anti-apoptotic molecular target for the treatment of B-lineage leukemias and lymphomas. Early preclinical and clinical findings suggest that BTK inhibitors are useful in the treatment of these malignancies and have applications in the prevention and treatment of thromboembolic complications and inflammatory disorders [2,3].
A large number of BTK inhibitors have been developed with diverse binding modes, including both reversible and irreversible inhibitors. These compounds can be classified by their ability to trigger sequestration of BTK residue Y551, an important determinant of potency against FcR signaling [4]. BTK inhibitors are increasingly replacing chemotherapy-based regimens, especially in patients with CLL and mantle cell lymphoma (MCL), and have also received approval for other conditions [5].
Tyrosine kinases, including BTK, are important mediators of signaling cascades involved in diverse biological processes like growth, differentiation, metabolism, and apoptosis. Aberrant tyrosine kinase activity is implicated in the pathophysiology of cancer, and selective tyrosine kinase inhibitors are a promising approach for innovative genome-based therapeutics [6,2,3]. This study aims to contribute to the ongoing efforts in identifying potential BTK inhibitors by exploring the binding profiles of natural compounds.
Materials and Methods
This study employed computational techniques to investigate the interactions between ligands and protein receptors. The primary software used for this purpose was Flare (Cresset), a computational tool designed for ligand and structure-based drug discovery.
Flare facilitates the optimization and prioritization of new molecules through high-resolution 3D visualization and in-depth analysis of target structures and potential ligands. It enables users to closely examine ligand-protein complexes, providing insights into protein targets and ligand series. Flare supports both ligand- based and structure-based drug design to aid in lead optimization.
The core methodologies employed in this study were molecular docking and electrostatic complementarity analysis.
Molecular docking is a computational technique used to predict the binding orientation of a ligand (small molecule) to a protein receptor. It aims to identify the optimal conformation and orientation of the ligand within the protein's binding site, along with an estimate of the binding affinity. This method is widely employed in drug discovery to screen compound libraries, understand protein-ligand interactions, and guide the design of novel therapeutics.
Electrostatic complementarity analysis evaluates the interactions between molecules and their receptors. Electrostatic interactions are crucial for molecular recognition and are key determinants of binding free energy. Assessing the electrostatic matching in protein-ligand complexes provides insights into why ligands bind and how to improve binding. Ideally, the electrostatic potentials of the ligand and the protein at the protein-ligand interaction interface should exhibit a high degree of complementarity.
Parameter Definitions
To facilitate the interpretation of results, the following parameters are defined
• Rank Score: Optimized to accurately predict the 3D poses of
docked ligands.
• DG: Optimized to accurately estimate protein-ligand binding
energy, assuming correct ligand orientation.
• VS: Optimized for maximum efficiency in virtual screening experiments, discriminating between active and inactive compounds.
• Electrostatic Complementarity (EC): A normalized surface integral of the complementarity score, representing the average complementarity between the ligand and protein surfaces.
• EC r and EC rho: Pearson and Spearman correlation coefficients, respectively, calculated based on the electrostatic potentials of the ligand and protein.
All three scores range from -1 (perfect clash) to 1 (perfect complementarity). To simplify data presentation and interpretation, the negative sign of docking scores and free energy values was removed.
Natural Compound Selection and Preparation
Natural compounds included in this study were selected based on their presence in various plant species, as detailed in the pharmacognosy text by Bruneton [7]. The chemical structures of these compounds were retrieved from the PubChem database in SDF (Structure-Data File) format. These files were then used as input for the docking simulations performed with Flare (Cresset).
Results
Docking of BTK Inhibitor Drugs
This section presents a structure-based comparison between known BTK inhibitor drugs and Ibrutinib, utilizing the crystal structure of Bruton's tyrosine kinase with PDB ID 5P9J [3]. While BTK inhibitors have revolutionized the treatment of B-cell malignancies, challenges such as drug resistance, off-target effects, and varying toxicity profiles necessitate the continuous exploration of novel BTK inhibitors with improved pharmacological profiles.
The docking scores, particularly the ΔG values, indicate that evobrutinib and ibrutinib exhibit high binding affinities to BTK. Furthermore, these compounds also demonstrate favorable electrostatic complementarity, suggesting strong interactions with the target protein. It is important to acknowledge that while high binding affinity is crucial, it does not always translate to optimal clinical outcomes. For instance, ibrutinib, a first-generation BTK inhibitor, is associated with a range of side effects, including bleeding, atrial fibrillation, and arthralgias, which can limit its long-term use. Newer generation inhibitors, such as evobrutinib, aim to improve selectivity and reduce these off-target effects.
Computational studies such as the one presented here are essential for understanding the molecular interactions between inhibitors and BTK. These insights are crucial for guiding the development of more effective and safer therapeutic agents. The search for new molecules often involves exploring natural compounds as scaffolds, aiming to leverage their diverse structural features and potentially reduce toxicological profiles, ultimately leading to improved patient outcomes."
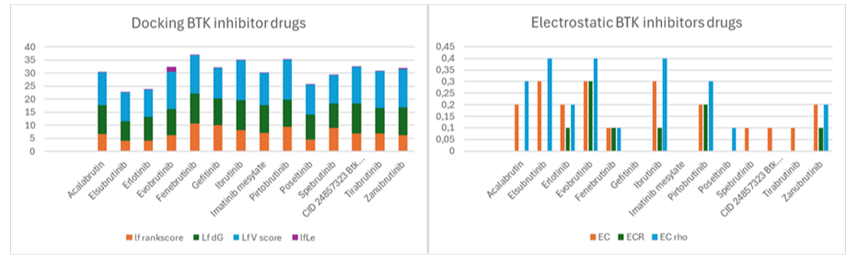
Figure 1: This figure presents the results of a computational docking study evaluating the binding affinities and electrostatic complementarity of various BTK inhibitors, To better understand the graphs look at the parameter definitions in materials and methods.
Docking of St. John's Wort Compounds on BTK
This section presents a structure-based comparison between Ibrutinib, a known BTK inhibitor, and compounds derived from St. John's wort (Hypericum perforatum), utilizing the crystal structure of Bruton's tyrosine kinase with PDB ID 5P9J [3]. The analysis focuses on both docking scores, reflecting binding affinity, and electrostatic complementarity, indicating favorable electrostatic interactions. The graphs provide a comparative overview of various compounds, highlighting their potential to interact with BTK.
The docking scores reveal that several Hypericum compounds exhibit strong binding affinities to BTK. Notably, hypericin, a polycyclic quinone derivative, demonstrates a high docking score, suggesting a robust interaction with the BTK binding site. Hyperforin, a phloroglucinol derivative, also shows promising docking scores, potentially due to its ability to interact with hydrophobic pockets within the BTK active site. Pseudohypericin, another polycyclic quinone, and amentoflavone, a biflavonoid, further contribute to the observed binding affinities. These compounds, with their diverse chemical structures, highlight the potential of St. John's wort as a source of BTK inhibitors.
It is worth noting that the efficacy of Hypericum compounds might be linked to their ecological role in the plant's survival. St. John's wort, when grazed by ruminants, relies on the passage of its seeds through the animal's digestive system for propagation. The compounds present in the plant, such as those described above, likely play a role in modulating the animal's gut environment. By attenuating inflammation and regulating gastric acid secretion, these compounds may create a more favorable environment for seed survival and germination. This evolutionary adaptation could explain the high efficacy observed in the docking scores, as these compounds are designed to interact with biological systems on a large scale.
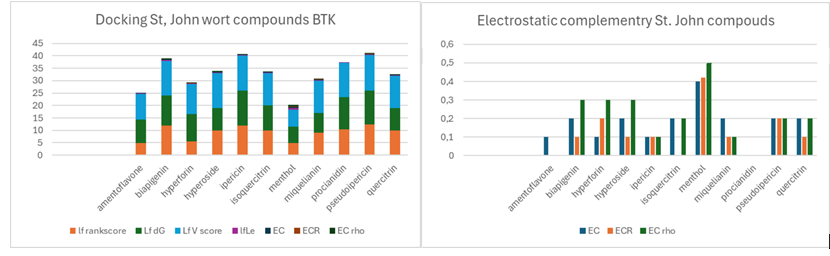
Figure 2: This figure presents the results of a computational docking study investigating the interactions between compounds derived from St. John's wort (Hypericum perforatum) and Bruton's tyrosine kinase (BTK), using the protein structure with PDB ID 5P9J. To better understand the graphs look at the parameter definitions in materials and methods.
Menthol presents a different scenario, as it is a molecule with reported anticancer activity [4]. While the precise mechanisms underlying these anticancer activities remain largely unknown, the high electrostatic affinity values observed in this study suggest a potential mechanism involving BTK inhibition.
The electrostatic complementarity (EC) values indicate that menthol exhibits a favorable fit within a specific region of the BTK binding pocket. Considering its known decongestant action, it is plausible that menthol influences BTK activity towards the modulation of metabolic pathways. The diverse orientations observed in docking studies suggest a flexible binding mode for menthol, allowing it to adapt to the pocket's contours. The range of electrostatic scores highlights the sensitivity of menthol's interaction to subtle conformational changes, demonstrating that even minor adjustments can significantly impact binding affinity. This variability underscores the importance of considering multiple conformers in docking studies to fully understand ligand- protein interactions.
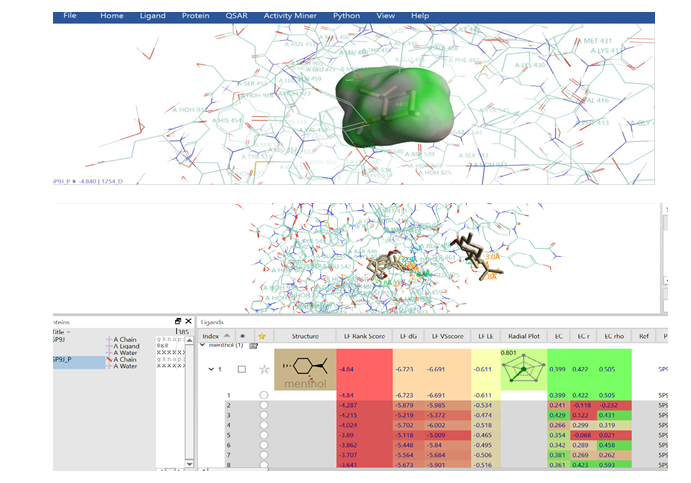
Figure 3: This figure showcases eight distinct conformations of menthol within the BTK binding pocket, each exhibiting varying degrees of docking and electrostatic affinity. The diverse orientations suggest a flexible binding mode for menthol, allowing it to adapt to the pocket's contours. To better understand the graphs look at the parameter definitions in materials and methods.
Docking of Compound Library (2632 Molecules) [8]
The following describes compounds within the library that exhibit docking scores similar to or higher than the reference drugs.
A structure-based comparison was performed between Ibrutinib and compounds from the library, utilizing the crystal structure of Bruton's tyrosine kinase with PDB ID 5P9J. Notably, established drugs such as docetaxel and simvastatin demonstrate high docking scores, indicating strong binding affinities. Among natural compounds, ginsenoside demonstrates significant electrostatic complementarity, suggesting favorable interactions with the target. Punicalagin and pentagalloylglucose, found in pomegranate (Punica granatum), also show promising electrostatic scores. Additionally, salvianolic acid from Salvia species presents notable docking and electrostatic profiles, highlighting the potential of these natural compounds for further investigation.
This library of compounds contains known drugs and natural compounds with established toxicological profiles. The repurposing of drugs already known for different pathologies (off-label use) can potentially save time and resources typically required for testing new molecules, as the testing of these drugs is already complete.

Figure 4: The presented graphs illustrate the docking and electrostatic complementarity scores of compounds from the library, including known drugs and natural molecules. For a better understanding of the graphs, refer to the parameter definitions in the Materials and Methods section.
Milk thistle, derived from various Old World thorny shrubs and subshrubs, including Silybum marianum, contains the active chemical component silymarin. Silymarin is a complex of flavonoids such as silibinin, dehydrosilybinin, silychristin, and silidianin. These compounds are known for their antioxidant properties and their ability to alter the structure of liver cell membranes, which can block the absorption of toxins and stimulate the production of new liver cells. Furthermore, milk thistle can increase cellular levels of adenosine triphosphate (ATP), demonstrating dose-dependent cytoprotection of cardiac myocytes against doxorubicin. Notably, silibinin, a component of milk thistle, has been shown to inhibit mitogenic and cell survival-mediated signaling by growth factor receptors, thus inhibiting tumor growth.
In the context of the current study, while the graphs illustrate the docking and electrostatic complementarity profiles of various compounds, including known drugs and natural molecules, it is important to note that milk thistle components, such as silibinin, also exhibit interesting profiles. This suggests that these compounds may also interact with the target protein in a manner that warrants further investigation. Note that adrenaline has a high electrostatic complementarity on BTK proteins, and this is plausible, as it is released by the adrenal glands in cases of fight or flight, and an anti-inflammatory effect allows in cases of emergency to give
less weight to pain and inflammation to facilitate escape in cases of emergency. From the screening of the compound library, it emerged that many compounds obtained scores comparable to BTK inhibitor drugs, and in particular, the anthraquinone compounds.
Let's analyze the quinone compounds contained in some plant species. From the Brunetton pharmacognosy text (reference 2), from the chapter on quinone compounds and related species, Rumex and rhubarb were found to be the species with quinone compounds at comparable scores. From St. John's wort (Hypericum perforatum) and rhubarb (Rheum rhabarbarum L)
Docking of Quinone-Containing Species
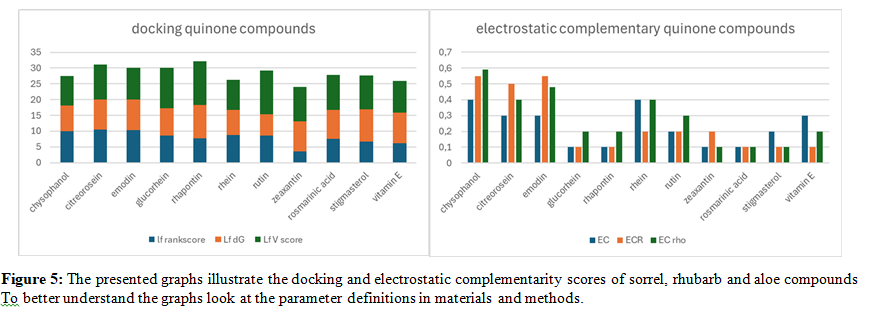
This section focuses on the docking analysis of compounds from plant species known to contain significant amounts of quinones, including Rumex (sorrel), rhubarb (Rheum rhabarbarum), and aloe (Aloe vera). Quinones are a class of organic compounds with diverse biological activities, and their presence in these plants suggests potential therapeutic applications.
The docking scores reveal that several quinone compounds from these species exhibit notable binding affinities to BTK. Specifically, compounds such as chrysoeriol and emodin demonstrate strong interactions, as evidenced by their high docking scores. Furthermore, the electrostatic complementarity analysis highlights favorable electrostatic interactions, particularly for these compounds. Rumex species, commonly known as sorrel, contain a variety of quinone derivatives, including chrysophanol and physcion. These compounds have been traditionally used for their laxative and anti-inflammatory properties. The docking scores suggest that these compounds may also interact with BTK, potentially contributing to their therapeutic effects.
Rhubarb (Rheum rhabarbarum) is another rich source of quinones, including rhein and aloe-emodin. These compounds are known for their purgative and anti-tumor activities. The docking analysis indicates that these compounds can effectively bind to BTK, suggesting a potential role in modulating B-cell signaling. Aloe (Aloe vera) contains emodin, a compound known for its anti- inflammatory and anti-cancer properties. Emodin has been shown to inhibit various inflammatory pathways, including the NF-κB pathway, which plays a crucial role in B-cell activation. The high docking scores of emodin with BTK suggest that it may also exert its anti-inflammatory effects through BTK inhibition.
The anti-inflammatory properties of emodin are particularly noteworthy. Emodin has been shown to reduce inflammation in various experimental models, including colitis and arthritis. Its ability to interact with BTK suggests that it may also modulate B-cell-mediated inflammation. These findings highlight the potential of quinone compounds from Rumex, rhubarb, and aloe as BTK inhibitors.
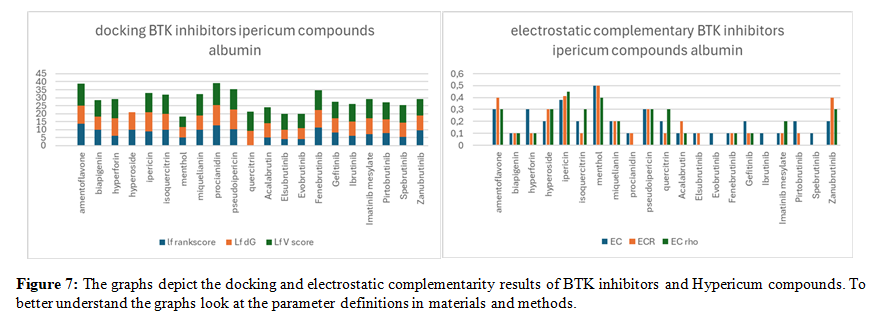
Albumin Binding Competition between BTK Inhibitors and St. John's Wort Compounds
Serum albumin plays a crucial role as the principal transporter for a wide array of drugs within the bloodstream. However, the binding sites on albumin are not exclusive to pharmaceuticals; they are also targeted by various biomolecules. This competition for binding sites can lead to the displacement of drugs, potentially altering their pharmacokinetics and efficacy. Therefore, understanding these competitive interactions is essential for predicting drug behavior and designing effective therapeutic strategies.
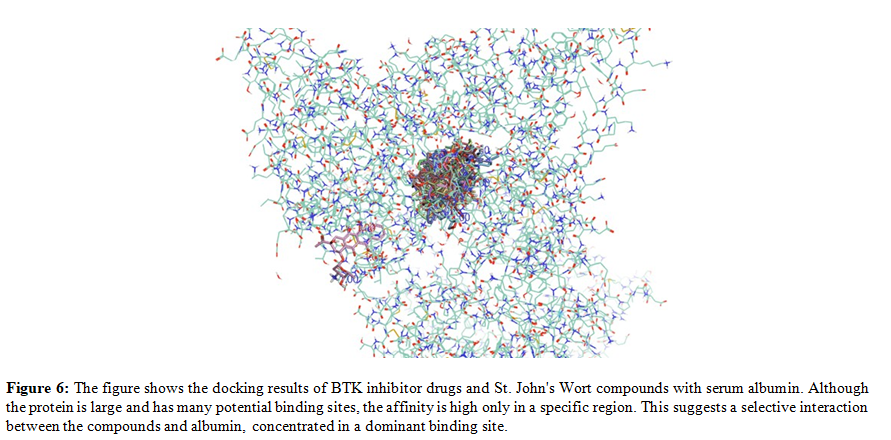
In this study, human albumin complexed with hydrarubicin (a quinone with a high affinity for BTK) was used as a template for docking calculations (PDB ID: 4LB2). Despite serum albumin being a globular protein of 69,000 Da, the docking results of BTK inhibitor drugs and Hypericum compounds, using a grid that covers almost the entire protein, demonstrate high affinity for only a specific portion of the protein, as illustrated in Figure n. 6.
Figure n. 6 shows the docking results of BTK inhibitor drugs and St. John's Wort compounds with serum albumin. Although the protein has a large surface area and many potential binding sites, the affinity is high only in a specific, concentrated region, suggesting a selective interaction between the compounds and a dominant binding site on albumin. Furthermore, several Hypericum compounds exhibit a higher affinity for serum albumin compared to certain BTK inhibitor drugs. This suggests that the concomitant use of these drugs with herbal products, such as Hypericum, rhubarb, or aloe, could potentially lead to the displacement of the drugs from their albumin binding sites. Such displacement may result in transient increases in drug plasma concentrations, potentially leading to temporary overdosing. Consequently, caution is advised when combining BTK inhibitors with herbal remedies, as competitive binding to serum albumin could significantly impact drug pharmacokinetics and overall safety several Hypericum compounds exhibit a higher affinity for serum albumin compared to certain BTK inhibitor drugs. This suggests that concomitant use of these drugs with herbal products like Hypericum, rhubarb, or aloe could potentially lead to the displacement of the drugs from their albumin binding sites.
Such displacement might result in transient plasma peaks of the drugs, leading to temporary overdosing. Therefore, caution is advised when combining BTK inhibitors with herbal remedies, as competitive binding to serum albumin could significantly impact drug pharmacokinetics and safety.
Conclusion
This study has explored the binding profiles of natural compounds and BTK inhibitors using computational docking and electrostatic complementarity analyses. It is important to critically evaluate the nature of docking and electrostatic complementarity scores: docking provides numerical values for the affinity of molecules towards the active site, but it does not guarantee the success of a molecule as a drug. In pharmacodynamic evaluations (drug-receptor interaction), docking scores are useful, but for pharmacokinetic evaluations, i.e., the ADME profile (absorption, distribution,
metabolism, and excretion), other parameters are necessary, such as TPSA (topological polar surface area), which can predict the behavior of passage or non-passage through membranes (for example, the blood-brain barrier). Docking also does not take into account induced fit; native protein ligands trigger mechanisms in receptor proteins that exclude other molecules from accessing the binding pockets, and this is unfortunately not reflected in docking scores.
The results highlight the potential of several natural compounds as BTK modulators, demonstrating significant binding scores and favorable electrostatic interactions. Notably, compounds from St. John's wort, Rumex, rhubarb, and aloe exhibited promising interactions with BTK, suggesting their potential therapeutic applications. High scores were also highlighted for quinone and anthraquinone compounds in particular, naturally present in different species such as St. John's wort, species of the Rumex genus such as bitter dock and rhubarb, and aloe.
Furthermore, the analysis of albumin binding competition revealed that certain Hypericum compounds exhibit a higher affinity for serum albumin compared to some BTK inhibitor drugs. This finding underscores the importance of considering drug-herb interactions, as concomitant use of herbal remedies could potentially lead to drug displacement and altered pharmacokinetics.
It is important to acknowledge that while computational studies provide valuable insights, they do not replace experimental validation. Further in vitro and in vivo studies are necessary to confirm these findings and explore the therapeutic potential of the identified natural compounds. It is also worth noting that research on natural compounds often takes a backseat to the development of synthetic molecules. This is largely due to the greater potential for financial remuneration associated with synthetic drug development. However, natural compounds offer a rich source of diverse chemical structures and biological activities, and their exploration could lead to the discovery of novel therapeutic agents [9,10].
Future Perspectives
"The findings of this study highlight the promising potential of natural compounds as modulators of Bruton's tyrosine kinase (BTK). Several natural products, particularly those from Hypericum perforatum, Rumex species, Rheum rhabarbarum, and Aloe vera, demonstrated significant binding affinities and favorable electrostatic complementarity to BTK. Moving forward, these natural compounds, especially those exhibiting high docking scores, could serve as valuable scaffolds for the development of novel synthetic BTK inhibitors. The diverse chemical structures of these natural products offer a rich source of inspiration for medicinal chemists seeking to design compounds with improved potency, selectivity, and pharmacokinetic profiles.
Specifically, compounds such as hypericin, hyperforin, emodin, and others identified in this study could be subjected to structural modifications and optimization through synthetic chemistry. This approach could lead to the creation of novel BTK inhibitors with enhanced therapeutic potential, while potentially minimizing off- target effects and improving drug-like properties. Furthermore, future research should focus on validating these in silico findings through in vitro and in vivo studies. These studies would provide crucial information on the efficacy and safety of these natural compounds and their derivatives as BTK inhibitors. In conclusion, the exploration of natural compounds as scaffolds for drug development represents a promising avenue for the discovery of novel BTK inhibitors. This approach could lead to the development of more effective and safer therapies for hematological malignancies and other diseases associated with BTK dysregulation."
Statement on the use of Generative Artificial Intelligence
This manuscript has been carefully prepared, with the assistance of generative artificial intelligence tools. Gemini was specifically used for translation and grammatical correction. The authors affirm their responsibility for the content, integrity, and scientific accuracy of the work.
Funding
This study project was self-funded.
Acknowledgements
The author acknowledges the academic license of Flare provided by Cresset. This did not influence the design, execution, or interpretation of the study. The author declares no other conflicts of interest
Software Used
The graphs were made with Microsoft Excel, academic license.
References
1. Patel, N. M., Oliveira, F. R., Ramos, H. P., Aimaretti, E.,Alves, G. F., Coldewey, S. M., ... & Thiemermann, C. (2023). Inhibition of Bruton’s tyrosine kinase activity attenuates hemorrhagic shock-induced multiple organ dysfunction in rats. Annals of Surgery, 277(3), e624-e633.
2. Uckun, F. M., & Qazi, S. (2010). Bruton's tyrosine kinase as a molecular target in treatment of leukemias and lymphomas as well as inflammatory disorders and autoimmunity. Expert Opinion on Therapeutic Patents, 20(11), 1457-1470.
3. Zhao, Y., Pan, H., Liu, W., Liu, E., Pang, Y., Gao, H., ... & Guo, J. (2023). Menthol: An underestimated anticancer agent. Frontiers in pharmacology, 14, 1148790.
4. 5P9J BTK1 COCRYSTALLIZED WITH IBRUTINIB. (n.d.).PDB.
5. Burger, J. A. (2019). Bruton tyrosine kinase inhibitors: present and future. The Cancer Journal, 25(6), 386-393.
6. Paul, M. K., & Mukhopadhyay, A. K. (2004). Tyrosine kinase–role and significance in cancer. International journal of medical sciences, 1(2), 101.
7. Bruneton, J. (2016). Pharmacognosie Phytochimie Plantes medicinales. Lavoisier. ISBN-13 f : f 978-2852069114
8. Selleckchem. (n.d.). Cinchonidine.
9. Bauer, M. R., & Mackey, M. D. (2019). Electrostatic complementarity as a fast and effective tool to optimize binding and selectivity of protein–ligand complexes. Journal of medicinal chemistry, 62(6), 3036-3050.
10. Bender, A. T., Gardberg, A., Pereira, A., Johnson, T., Wu, Y., Grenningloh, R., ... & Liu-Bujalski, L. (2017). Ability of Bruton’s tyrosine kinase inhibitors to sequester Y551 and prevent phosphorylation determines potency for inhibition of Fc receptor but not B-cell receptor signaling. Molecular pharmacology, 91(3), 208-219.