


Research Article - (2021) Volume 6, Issue 1
Clinical Significance of Spot Urinary Chloride Concentration Measurements in Patients with Acute Heart Failure: Investigation on the Basis of the TubuloGlomerular Feedback Mechanism
Received Date: Apr 10, 2021 / Accepted Date: Apr 07, 2021 / Published Date: Apr 25, 2021
Copyright: ©Hajime Kataoka,. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Citation: Hajime Kataoka (2021) Clinical Significance of Spot Urinary Chloride Concentration Measurements in Patients with Acute Heart Failure: Investigation on the basis of the Tubulo-Glomerular Feedback Mechanism. Cardio Open, 6(1): 115-123.
Abstract
Urinary chloride (Cl) is the key electrolyte for regulating renin secretion at the macula densa under the ‘tubulo-glomerular feedback’. Whether or not Cl filtrated into the urinary tubules actually associates with plasma renin activity (PRA) in clinical heart failure (HF) remains unclear. Data from 29 patients with acute worsening HF (48% men; 80.3±12 years) were analyzed. Blood and urine samples were immediately obtained before decongestive therapy after the patients rested in a supine position for 20-min. Clinical tests included peripheral blood tests, serum and spot urinary electrolytes, b-type natriuretic peptide (BNP), plasma neurohormones, and fractional urinary electrolyte excretion. In the 29 patients, urinary Cl concentrations inversely correlated with logarithmically transformed PRA (R2 =0.41, p=0.0002). The correlation was weaker in worsening chronic HF patients (R2 =0.32, p=0.01) compared with de novo HF patients (R2 =0.70, p=0.0026). Patients were divided into 2 groups according to the median urinary Cl concentration, a low group and a high group. Compared with the high group (100~184 mEq/L; n=14), the low group (4~95 mEq/L; n=15) exhibited more renal (serum creatinine; 1.45±0.63 vs 1.00±0.38 mg/d, p=0.029) and cardiac (log BNP; 2.99±0.3 vs 2.66±0.32 pg/mL, p=0.008 p=0.008) impairment, and higher PRA (3.42±4.7 vs 0.73±0.46 ng/mL/h, p=0.049), and lower fractional excretion of urinary Cl (1.34±1.3 vs 5.33±4.1%, p<0.0001). The present study provides clinical data on the possible functioning of urinary Cl involved in the mechanism of ‘tubulo-glomerular feedback’, and thus advances our understanding of the clinical meanings of the significance of urinary Cl concentration measurement.
Keywords
Acute Heart Failure, Chloride, Chloride Theory, Urinary Chloride Concentration, Tubulo-Glomerular Feedback, Plasma Renin Activity
Introduction
In the kidney, salt sensing, renin secretion, and ‘tubulo-glomeru- lar feedback’ are dependent on Chloride (Cl) rather than Sodium (Na) [1-5]. Despite mounting experimental and clinical data on the neurohormonal activity occurring during heart failure (HF), actual clinical data clarifying the role of urinary Cl, which is pos- sibly involved in ‘tubulo-glomerular feedback’, is lacked [6-12]. Urinary electrolyte analysis, particularly urinary Na, was recently recognized to be important for the evaluation, treatment, and prog- nosis of clinical HF pathophysiology [13-23]. Analysis of urinary Cl may contribute to our understanding the role of the electrolyte Cl in ‘tubulo-glomerular feedback’ because Cl, not Na, is the key electrolyte for regulating renin secretion. To the best of my knowl- edge, however, there are hardly any clinical studies examined as- sociation between urinary Cl concentration and plasma renin ac- tivity (PRA) to provide evidence for ‘tubulo-glomerular feedback’ actually functioning in the human body.
Thus, the present study aimed to determine the relation of the uri- nary Cl concentration to PRA in patients with acute HF in an effort to clarify the function of Cl in the ‘tubulo-glomerular feedback’. Moreover, the clinical significance of spot urinary Cl testing in pa- tients with acute HF was evaluated according to the urinary Cl-cen- tered ‘tubulo-glomerular feedback’ model. The present study also sought to provide clinical evidence supporting the ‘chloride theo- ry’ for HF pathophysiology, which states that changes in the serum Cl concentration are deeply associated with changes in plasma volume, hemodynamics, and neurohormonal activity [12, 24, 25].
Methods
Study Design
This study was a retrospective single-center observational study that enrolled 31 consecutive patients with acute HF at Nishida Hospital (Saiki-city, Oita, Japan) participating in a neurohormonal study between March 2017 and April 2018. Diagnosis of worsen- ing HF was established by standard clinical criteria according to presentation, echocardiography, and serum b-type natriuretic pep- tide (BNP) [26]. Additional routine tests included thoracic ultra- sound to evaluate the presence of pleural effusion [27,28]. Wors- ening HF was treated by conventional therapy with a combination of loop diuretics, aldosterone blockade, thiazide diuretics, oral va- sopressin antagonist, acetazolamide, and/or inotropic drugs by oral and/or intravenous routes in the hospital or outpatient clinic. Based on the follow-up examination, the response of worsening HF to treatment and the return of the clinical presentation to stable HF status were determined. Acute HF patients with cardiogenic shock, clinical diagnosis of acute coronary syndrome, known advanced renal disease (serum creatinine level >3.0 mg/dL) were excluded from the study.
Data Collection and Analytic Methods
Physical examination, peripheral venous blood tests (hematologic, BNP, and neurohormonal tests), and a spot urine test were per- formed at presentation of the acute HF episode immediately before the initiation of decongestive therapy. The blood and urine sam- ples were obtained after patients rested in a supine or semi-supine position for 20-min. Peripheral blood tests, analyzed by standard techniques, included hemoglobin, hematocrit, serum electrolytes (Na, K, and Cl), blood urea nitrogen, and creatinine. The spot urine test included measurement of electrolyte and creatinine concentra- tions, and osmolality. Plasma BNP was measured by chemilumi- nescent immunoassay. Plasma adrenaline and noradrenaline lev- els were measured by high-performance liquid chromatography. Plasma renin activity was measured by enzyme immunoassay. Plasma aldosterone and arginine vasopressin (AVP) levels were measured by radioimmunoassay. Fractional electrolyte excretions were calculated as: fractional excretion of X = (Xurine/Xserum)×(Crse- rum/Crurine)×100 [29,30]. Urinary osmotic pressure was measured by the freezing point depression method using an OM-6060 type automatic osmotic pressure measuring device (Arkray Inc., Kyoto, Japan).
Statistical Analysis
All data are expressed as mean ± SD for continuous data and per- centage for categorical data. Paired and unpaired t tests for con- tinuous data and Fisher’s exact test for categorical data were used for 2-group comparisons. Pearson’s correlation was performed to evaluate the linear association between logarithmically trans- formed PRA and other variables. A p value of <0.05 was consid- ered statistically significant.
Results
Of the 31 acute HF patients, 2 were excluded from the present study due to insufficient clinical data. The remaining 29 patients (48% men; 80.3±12 years), including de novo acute HF patients (n=10), were enrolled in the analysis. Clinical characteristics of the study patients at baseline are shown in Table 1. The primary causes of worsening HF varied; atrial fibrillation was observed in 14 (48%) patients. All study patients presented with 2 to 4 HF signs on the basis of physical examination and evaluation of pos- sible pleural effusion by thoracic ultrasound. Plasma BNP levels were elevated: definitely (≥500 pg/mL) in 20 patients, moderately (200 pg/mL to <500 pg/mL) in 7, and mildly (100 pg/mL to <200 pg/mL) in 2. No diuretics were prescribed for 10 patients with de novo HF episode before current presentation to the hospital. Of the 29 patients, 24 were treated for acute HF in the hospital, and 5 were treated at the outpatient clinic.
Table 1: Clinical characteristics of the study patients
|
Characteristics |
Total (N = 29) |
|
Age (years) |
|
|
Mean ± SD |
80.3±12 |
|
Range |
53-97 |
|
Male |
14 (48) |
|
Primary cause of HF |
|
|
Hypertension |
19 (64) |
|
Valvular |
6 (22) |
|
Ischemic/Cardiomyopathy |
3 (11) |
|
Arrhythmia |
1 (3) |
|
Left ventricular EF (%) |
|
|
Mean ± SD |
47.8±18 |
|
Left ventricular EF > 50% |
15 (52) |
|
Atrial fibrillation |
14 (48) |
|
NYHA-FC |
|
|
III |
6 (21) |
|
IV |
23 (79) |
|
HF-related physical findings |
|
|
Bilateral leg edema around or above the ankle |
24 (84) |
|
Bilateral pulmonary rales beyond the basal lung |
22 (76) |
|
Pleural effusion on thoracic ultrasound |
25 (86) |
|
Third heart sound (S3) |
5 (17) |
|
Number of HF signs (mean ± SD; range) |
2.7±0.6; 2¬Ã¢??4 |
|
B-type natriuretic peptide (pg/mL) |
|
|
2000≥ |
1 (3) |
|
2000 – 1000 |
7 (24) |
|
1000 – 500 |
12 (42) |
|
500 – 200 |
7 (24) |
|
200 – 100 |
2 (7) |
|
Baseline medication use |
|
|
De novo HF patients without diuretic treat- ment |
10 (34) |
|
Diuretics |
|
|
Loop diuretics |
13 (45) |
|
Thiazide diuretics |
4 (14) |
|
MRA |
12 (42) |
|
Tolvaptan |
6 (21) |
|
ACE inhibitors/ARB |
7 (24) |
|
Beta-blockers |
9 (31) |
|
Calcium antagonists |
12 (42) |
|
Digitalis |
3 (11) |
|
Nitrates |
2 (7) |
Data presented as number (%) of patients otherwise specified. ACE: angiotensin-converting enzyme, ARB: angiotensin II receptor blocker, EF: ejection fraction, MRA: mineralocorticoid receptor antagonist, NYHA-FC: New York Heart Association functional class, HF: heart failure. Pearson’s correlation of Log PRA (ng/mL/h) with multiple vari- ables under acute HF (n = 29) is shown in Table 2. The urinary Cl and Na concentrations each inversely correlated well with the Log PRA in a total of 29 HF patients (Cl, R2=0.41, p=0.0002, Fig- ure 1A; Na, R2=0.54, p=<0.00001). The effects of cardiovascular medication (Table 3) on the relationship between the plasma renin activity and urinary Cl concentration are shown in Figures 1B and 1C, in which the correlation between them was determined sepa- rately in worsening chronic HF patients (Figure 1B; n=19; more cardiovascular-related medication) and de novo HF patients (Fig- ure 1C; n=10; less cardiovascular-related medication). The cor- relation was weaker in worsening chronic HF patients (R2=0.32, p=0.01, Figure 1B) compared with de novo HF patients (R2=0.70, p=0.0026, Figure 1C).
Table 2: Pearson’s correlation of log PRA (ng/mL/h) with multiple variables under acute heart failure (Total N = 29)
|
Variable |
R2 |
p-value |
|
Systolic BP (mmHg) |
0.07 |
0.17 |
|
Diastolic BP (mmHg) |
0.001 |
0.86 |
|
Heart rate (beats/min) |
0.014 |
0.53 |
|
Serum electrolytes |
||
|
Sodium (mEq/L) |
0.024 |
0.42 |
|
Potassium (mEq/L) |
0.0003 |
0.93 |
|
Chloride (mEq/L) |
0.07 |
0.16 |
|
BUN (mg/dL) |
0.096 |
0.1 |
|
Creatinine (mg/dL) |
0.007 |
0.66 |
|
Log BNP (pg/mL) |
0.043 |
0.28 |
|
Urinary concentration |
||
|
Sodium (mEq/L) |
0.54 |
<0.0001* |
|
Potassium (mEq/L) |
0.19 |
0.017* |
|
Chloride (mEq/L) |
0.41 |
0.0002* |
|
Osmolality (mOsm/kg H2O) |
0.023 |
0.43 |
|
Adrenaline (pg/mL) |
0.022 |
0.44 |
|
Noradrenaline (pg/mL) |
0.21 |
0.013* |
|
Aldosterone (pg/mL) |
0.43 |
0.0001* |
|
AVP (pg/mL) |
0.02 |
0.46 |
AVP: arginine vasopressin, BNP: b-type natriuretic peptide, BP: blood pressure, BUN: blood urea nitrogen, PRA: plasma renin activity.
*Statistically significant (p < 0.05).
Table 3: Comparison of the baseline medication use between worsening chronic HF patients and de novo HF patients (Total N = 29)
|
|
Worsening chronic HF pa- tients (n = 19) |
De novo HF patients (n = 10) |
p-value |
|
|
Diuretics |
19 (100) |
0 |
<0.0001* |
|
|
Loop diuretics |
13 (68) |
0 |
<0.0001* |
|
|
Thiazide diuretics |
4 (21) |
0 |
0.27 |
|
|
MRA |
12 (63) |
0 |
0.001* |
|
|
Tolvaptan |
6 (32) |
0 |
0.07 |
|
|
Acetazolamide |
9 (47) |
0 |
0.01* |
|
|
Neurohormonal blockades |
13 (68) |
2 (20) |
0.01* |
|
|
ACE inhibitors/ARB |
5 (26) |
2 (20) |
1 |
|
|
Beta-blockers |
8 (42) |
1 (10) |
0.1 |
|
|
|
1 drug of above |
13 (68) |
1 (10) |
0.005* |
|
|
2 drugs of above |
0 |
1 (10) |
0.34 |
Data presented as number (%) of patients. ACE: angiotensin-converting enzyme, ARB: angiotensin II receptor blocker, MRA: mineralo-corticoid receptor antagonist. *Statistically significant (p <0 .05, Fisher’s exact test).
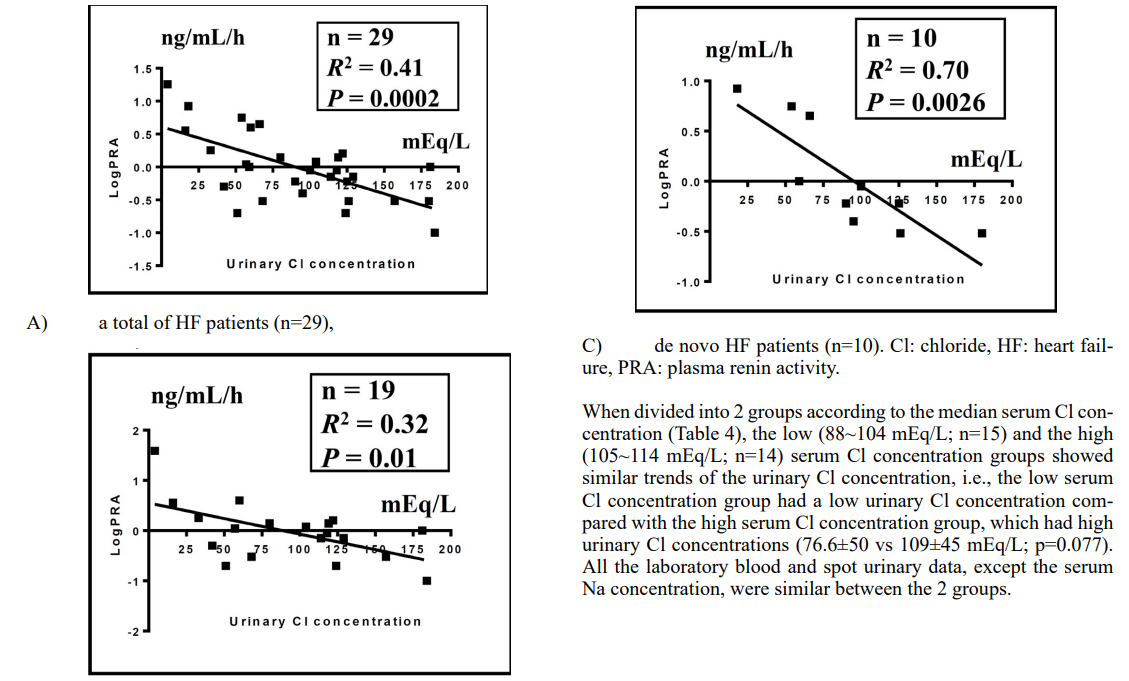
Table 4: Comparisons of low and high groups stratified on the basis of the serum chloride concentration, urinary chloride con- centration, or plasma renin activity in 29 patients with acute heart failure
|
|
Total (N=29) |
Serum Chloride Concentration (mEq/L) |
Urinary Cl Concentration (mEq/L) |
Plasma Renin Activity (ng/mL/h) |
||||||
|
Low (n=15) |
High (n=14) |
p-value |
Low (n=15) |
High (n=14) |
p-value |
Low (n=15) |
High (n=14) |
p-value |
||
|
88 ~ 104 |
105 ~ 114 |
4 ~95 |
100 ~ 184 |
0.1 ~ 0.9 |
1.0 ~ 18 |
|||||
|
Vital signs |
||||||||||
|
Systolic BP (mmHg) |
137±35 |
136±31 |
139±39 |
0.86 |
144±36 |
130±33 |
0.3 |
144±37 |
130±32 |
0.29 |
|
Diastolic BP (mmHg) |
79±19 |
77±18 |
81±25 |
0.56 |
79±17 |
79±21 |
0.98 |
78±20 |
80±18 |
0.75 |
|
Heart rate (beats/ min) |
86.5±21 |
84.3±18 |
88.8±25 |
0.58 |
87.5±20 |
85.3±23 |
0.78 |
81.9±25 |
91.3±15 |
0.24 |
|
Laboratory blood data |
||||||||||
|
Hemoglobin (g/dL) |
11.8±2.1 |
11.9±2.2 |
11.8±2.2 |
0.91 |
11.8±2.2 |
11.9±2.1 |
0.92 |
11.8±2.1 |
11.9±2.2 |
0.89 |
|
Hematocrit (%) |
35.9±6.4 |
35.8±6.9 |
35.9±6.2 |
0.95 |
35.7±6.8 |
36.0±6.2 |
0.92 |
35.6±6.2 |
36.2±6.9 |
0.8 |
|
Serum electrolytes |
||||||||||
|
Sodium (mEq/L) |
139±4.8 |
136±4.6 |
142±3.1 |
0.0007* |
138±4.9 |
139±4.8 |
0.37 |
138±6.0 |
139±3.3 |
0.66 |
|
Potassium (mEq/L) |
4.27±0.6 |
4.33±0.6 |
4.20±0.7 |
0.61 |
4.30±0.7 |
4.23±0.6 |
0.77 |
4.25±0.6 |
4.29±0.7 |
0.87 |
|
Chloride (mEq/L) |
103±6.2 |
98.8±4.8 |
108±3.5 |
<0.0001* |
102±7.1 |
104±5.1 |
0.38 |
103±6.1 |
103±6.6 |
0.99 |
|
BUN (mg/dL) |
27.0±13 |
26.4±14 |
27.6±11 |
0.8 |
31.4±13 |
22.2±10 |
0.047* |
24.3±12 |
29.8±13 |
0.25 |
|
Creatinine (mg/dL) |
1.23±0.56 |
1.24±0.53 |
1.23±0.62 |
0.99 |
1.45±0.63 |
1.00±0.38 |
0.029* |
1.18±0.58 |
1.29±0.56 |
0.6 |
|
Log BNP (pg/mL) |
2.83±0.35 |
2.88±0.40 |
2.77±0.29 |
0.42 |
2.99±0.30 |
2.66±0.32 |
0.008* |
2.81±0.26 |
2.85±0.43 |
0.76 |
|
Neurohormonal test |
||||||||||
|
Adrenaline (pg/mL) |
0.08±0.07 |
0.077±0.07 |
0.087±0.08 |
0.74 |
0.095±0.08 |
0.067±0.06 |
0.31 |
0.068±0.06 |
0.096±0.08 |
0.31 |
|
Noradrenaline (pg/ mL) |
1.02±0.67 |
1.15±0.69 |
0.88±0.63 |
0.28 |
1.24±0.78 |
0.78±0.43 |
0.72 |
0.76±0.38 |
1.29±0.8 |
0.03* |
|
Renin activity (ng/ mL/h) |
2.12±3.6 |
2.96±4.8 |
1.23±1.4 |
0.2 |
3.42±4.7 |
0.73±0.46 |
0.049* |
0.47±0.26 |
3.90±4.6 |
0.008* |
|
Aldosterone (pg/ mL) |
128±106 |
146±119 |
109±90 |
0.36 |
158±121 |
96.4±79 |
0.19 |
86.2±71 |
174±120 |
0.023* |
|
AVP (pg/mL) |
3.55±3.5 |
3.15±2.7 |
3.97±4.2 |
0.53 |
4.17±4.1 |
2.89±2.6 |
0.11 |
2.95±2.6 |
4.19±4.2 |
0.34 |
|
Laboratory urinary data (spot urine) |
||||||||||
|
Concentration of urinary electrolytes |
||||||||||
|
Sodium (mEq/L) |
90.8±46 |
80.1±48 |
102±43 |
0.21 |
56.7±30 |
127±30 |
<0.0001* |
113±37 |
66.9±44 |
0.005* |
|
Potassium (mEq/L) |
30.8±18 |
31.5±20 |
30.1±16 |
0.84 |
36.7±22 |
24.5±9.2 |
0.006* |
26.3±14 |
35.6±21 |
0.17 |
|
Chloride (mEq/L) |
92.3±50 |
76.6±50 |
109±45 |
0.077 |
52.9±27 |
135±29 |
<0.0001* |
114±41 |
70.0±49 |
0.01* |
|
% excretion of urinary electrolytes |
||||||||||
|
Sodium (%) |
2.45±2.6 |
2.47±2.5 |
2.42±2.8 |
0.95 |
1.15±1.3 |
3.83±3.0 |
<0.0001* |
2.94±2.8 |
1.92±2.3 |
0.3 |
|
Potassium (%) |
16.4±9.5 |
17.6±11 |
15.1±8.3 |
0.49 |
14.0±8.9 |
19.1±9.7 |
0.006* |
15.7±7.4 |
17.2±12 |
0.69 |
|
Chloride (%) |
3.27±3.6 |
3.28±3.5 |
3.25±3.7 |
0.98 |
1.34±1.3 |
5.33±4.1 |
<0.0001* |
3.83±3.8 |
2.67±3.4 |
0.39 |
|
Osmolality (mOsm/ kg H2O) |
470±183 |
443±167 |
498±202 |
0.43 |
487±198 |
451±171 |
0.014* |
465±180 |
474±194 |
0.9 |
AVP: arginine vasopressin, BNP: b-type natriuretic peptide, BP: blood pressure, BUN: blood urea nitrogen. *Statistically significant difference be- tween before and after treatment (p < 0.05, unpaired t test).
When divided into 2 groups based on the median urinary Cl con- centration (Table 4), the low urinary Cl concentration group (4~95 mEq/L; n=15) exhibited more renal (serum creatinine; 1.45±0.63 vs 1.00±0.38 mg/d, p=0.029) and cardiac (log BNP; 2.99±0.30 vs 2.66±0.32 pg/mL, p=0.008) impairment compared with the high urinary Cl concentration group (100~184 mEq/L; n=14). The low urinary Cl concentration group also exhibited higher PRA (3.42±4.7 vs 0.73±0.46 ng/mL/h, p=0.049), and a lower fractional excretion of urinary Cl (1.34±1.3 vs 5.33±4.1%, p<0.0001), Na (1.15±1.3 vs 3.83±3.0%; p<0.0001), and K (14.0±8.9 vs 19.1±9.7%; p=0.006). The plasma AVP level tended to be high in the low urinary Cl con- centration group compared with the high urinary Cl concentration group (4.17±4.1 vs 2.89±2.6 pg/mL, p=0.1). Urine osmolarity was higher in the low urinary Cl concentration group than in the high group (487±198 vs 451±171 mOsm/kgH2O, p=0.014).
When divided into 2 groups according to the median PRA (Table 4), the high PRA group (1.0~18 ng/mL/h; n=14) had high serum values of noradrenaline (1.29±0.8 vs 0.76±0.38 pg/mL, p=0.03) and aldosterone (174±120 vs 86.2±71 pg/mL, p=0.023), and low spot urinary Cl (70.0±49 vs 114±41 mEq/L, p=0.01) and Na (66.9±44 vs 113±37 mEq/L, p=0.005) concentrations compared with the low PRA group (0.1~0.9 ng/mL/h; n=15).
Discussion
The present study, in which acute HF patients were analyzed im- mediately before initiating decongestive treatment (pre-treatment phase), provides clinical data on the possible functioning of ‘tubu- lo-glomerular feedback’ by evaluating the relation of spot urinary Cl concentration with PRA, which should advance our understand- ing of the clinical significance and utility of urinary electrolyte analysis, and also provides data supporting the concept of the ‘chloride theory’ of HF pathophysiology [12,24,25].
Function of ‘Tubulo-Glomerular Feedback’ in Acute HF
Many experimental studies have demonstrated the central role of urinary Cl in the ‘tubulo-glomerular feedback’ mechanism, through which absorption and excretion of urinary electrolytes and water along the urinary tubules are integrated for maintaining body fluid dynamics [1-4]. It remains unclear, however, whether the urinary Cl concentration regulates PRA in clinical HF patho- physiology of human body. Although the urine content, including electrolyte Cl, should be modified by the process of glomerular filtration of plasma running through the renal tubules to the blad- der, the present study revealed a modest correlation between the spot urinary Cl concentration and PRA (Figure 1), confirming the possible functioning of ‘tubulo-glomerular feedback’ in the human body. Indeed, the scatter plot of individual HF patients in the pres-ent study shown in Figure 1 is consistent with the findings reported by He et al, in which the relationship between the macula densa Cl concentration and renin secretion was evaluated in an isolated per- fused rabbit juxtaglomerular apparatus preparation [3]. Of course, the Cl concentration in the urine coming into the macula densa is not the only factor affecting the release of plasma renin.
The 4 main stimuli for renin release are:
(1) decreased baroreceptors stretch in afferent arterioles,
(2) decreased Na and Cl delivery to the macula densa,
(3) activation of renal sympathetic nerves and stimulation of b-ad- renergic receptors, and
(4) decreased negative feedback signaling through angiotensin II [5,31].
The present study provides important information regarding the effects of cardiovascular medication on the PRA and ‘tubulo-glo- merular feedback’ mechanism. As shown in Figure 1, the urinary Cl concentration was not well correlated with PRA in worsening chronic HF patients (Figure 1B) compared with de novo HF pa- tients (Figure 1C). The different correlation strengths between them would be influenced by the different types of cardiovascular medications in each population: no diuretic medication and fewer neurohormonal blockers (angiotensin-converting enzyme, angio- tensin II receptor, or beta blockers) in de novo HF patients, and vice versa in worsening chronic HF patients (Table 3). Indeed, it is well known that cardiovascular medications greatly affect re- nin-angiotensin-aldosterone system activity. Loop diuretics can stimulate renin release by inhibiting macula densa Na/Cl transport in the kidney, which mimics a situation of low sodium/chloride delivery to the macula densa and thus elicits renin secretion [5,32]. Angiotensin-converting enzyme and angiotensin II receptor block- ers inhibit angiotensin II-negative feedback, leading to an increase in PRA. In contrast, beta-blockers reduce renin levels by suppress- ing beta-adrenergic stimulation of the kidney [33]. Therefore, ex- treme caution is needed when evaluating the tubulo-glomerular feedback mechanism in patients receiving diuretics and neurohor- monal blockers.
Clinical Significance of the Spot Urinary Cl Concentra- tion Measurement
Many recent clinical studies, except several reports that examined urinary Na concentration before initiation of acute HF treatment (pre-treatment), have focused on investigating the characteristics of urinary electrolytes after diuretic treatment, particularly the uri- nary Na concentration or excretion, as an early marker for pre- dicting diuretic responsiveness and a long-term prognostic marker in patients with acute HF [13-23]. Almost exclusively, these stud- ies found that a low urinary Na concentration or excretion either pre- or post-diuretic treatment for worsening HF is associated with a poor short-term diuretic response and a high risk of long-term mortality. Most of these previous studies, however, did not con- sider urinary electrolyte levels as useful clinical information for understanding the complex HF pathophysiology and clarifying different HF profiles. Among these studies, Martens et al, examine serial urinary Na concentrations before and after an acute HF epi- sode, and offered novel insight into the potential mechanisms con- tributing to the developing of acute HF [23]. Similar to the study by Martens et al, in that the urinary Na concentration was inves- tigated, the present study demonstrated heterogenous distribution of the spot urinary Cl concentration and ability to phenotype acute HF status on the basis of this information, as described below [23].
Given the possible evidence for the actual functioning of ‘tubu- lo-glomerular feedback’ in acute HF patients reported here, it is expected that information obtained from a spot urinary Cl concen- tration can integrate the separate HF elements comprising individ- ual HF status (e.g., neurohormonal activities, electrolytes status, and renal and cardiac functions) into construction of systematic and reasonable linkages among them. As such, a pre-treatment spot urinary Cl test in acute HF patients would provide useful clin- ical data toward characterizing each acute HF phenotype before de novo diuretic treatment or adjustment of a recent diuretic regimen. That is, when acute HF patients were stratified into 2 groups on the basis of the median value of the pre-treatment urinary Cl con- centration, the low group exhibited more compromised renal and cardiac functions than the high group. More importantly, spot uri- nary Cl analysis provides data on differential activation of neuro- hormonal systems between groups under acute HF pathophysiol- ogy, including renin and AVP hormones (i.e., high neurohormonal activation in acute HF patients with a low urinary Cl concentration vs low neurohormonal activation in those with a high urinary Cl concentration), and provides essential information for dynamic movement of urinary Cl (also including Na, K, and water) [i.e., low (or high) urinary excretion of Cl in groups with low (or high) urinary Cl concentration under high (or low) levels of neurohor- monal activation]. The observed neurohormonal changes in renin and aldosterone were analogous to the study by Honda et al, that showed lower urinary Na concentration was associated with in- creased neurohormonal activities [22]. The present study suggests that acute HF patients with a low uri- nary Cl concentration in the pre-treatment test may be expected to have loop diuretic resistance because such an acute HF phenotype already exhibits compromised renal and cardiac functions under a highly activated neurohormonal system, as was similarly reported by Hanberg et al. [20]. Urinary Cl-centered information linked to the ‘tubulo-glomerular feedback’ model may be useful for decid- ing therapeutic options, because loop diuretic-resistant acute HF patients with a low urinary Cl concentration may respond well to treatment with Cl-regaining diuretics, such as acetazolamide, va- sopressin receptor antagonist, and sodium-glucose cotransporter-2 inhibitor [34]. Acute HF patients with low urinary Cl concentra- tion might be candidates for treatment by renin-angiotensin-aldo- sterone system blockade if there are no contraindications, such as coexisting renal dysfunction, hypotension, and HF with preserved ejection fraction [35].
Clinical Proof of the ‘Chloride Theory’ for HF Patho- physiology
Several years ago, a unifying hypothesis of the ‘chloride theory’ for HF pathophysiology was proposed, that states that changes in the serum Cl concentration is the primary determinant of chang- es in the plasma volume and neurohormonal activity under wors- ening HF and its resolution [24,25]. The proposed hypothesis is based on assumed interactions between changes in the serum Cl concentration and neurohormonal systems, but it has remained unclear whether this hypothesis is truly applicable to clinical re- al-world HF pathophysiology because previous clinical studies examined, without exception, the association of Na (not Cl) and water to the neurohormonal system [12]. Observations in the pres- ent study support the ‘chloride theory’ for HF pathophysiology on the basis of interactions between urinary Cl concentration and neurohormonal activities during worsening HF, i.e., renin activity becomes depressed in acute HF patients with an increased serum Cl concentration and renin activity becomes enhanced in those with a decreased serum Cl concentration [24,25]. Importantly, the present study clearly demonstrates that, in acute HF patients with a decreased urinary Cl concentration, excretion of Cl into the renal tubules is depressed, or, alternatively, their absorption from the renal tubules is enhanced, under high renin activation, but the serum Cl concentration does not increase effectively, probably due to insufficient supply of enough amount of Cl into the vascular or extracellular space, owing to yet unknown mechanisms. These observations have demonstrated the potential real-world applica- bility of the ‘chloride theory’ to clinical HF pathophysiology.
Study Limitations
This study was performed in a relatively small number of patients comprising a clinically heterogeneous population (e.g., both HF with preserved and reduced left ventricular ejection fraction; both established and de novo HF; and both pre-existing diuretic ther- apy and diuretic naive), was a single-center observational study, and should be considered hypothesis-generating. The present re- sults were derived from a population of patients with moderate HF. Therefore, the results of this study cannot be generalized to patients with more advanced HF. Further studies including a larger number of HF patients with various HF conditions are needed to better assess the clinical implications of measurements of urinary Cl concentration in HF pathophysiology.
Conclusion
The present study provides the data on possible actual working of the ‘tubulo-glomerular feedback’ in the human body by evaluating the relation between spot urinary Cl concentration and PRA. In this study, it appears that the association of urinary Cl and Na with many variables was highly similar, but strictly speaking, this does not mean that electrolyte Na can be substituted for electrolyte Cl when evaluating HF pathophysiology because renin secretion and ‘tubulo-glomerular feedback’ are dependent on Cl rather than Na [1-5], and their prognostic significance and pathophysiologic roles are quite different [36-39]. Thus, it is reasonable to closely inves- tigate the serum and urinary Cl dynamics, as well as the serum and urinary Na dynamics, in HF pathophysiology in future clinical studies.
References
- Kotchen TA, Luke RG, Ott CE, Galla JH, Whitescarver S (1983) Effect of chloride on renin and blood pressure re- sponses to sodium chloride. Ann Intern Med 98: 817-822.
- Lorenz JN, Weihprecht H, Schnermann J, Skøtt O, Briggs JP (1991) Renin release from isolated juxtaglomerular appara- tus depends on macula densa chloride transport. Am J Physiol 260: F486-F493.
- He X-R, Greenberg SG, Briggs JP, Schnermann J (1995) Ef- fects of furosemide and verapamil on the NaCl dependency of macula densa-mediated renin secretion. Hypertension 26: 137-142.
- Schnermann J (1998) Juxtaglomerular cell complex in the reg- ulation of renal salt excretion. Am J Physiol 274: R263-R279.
- Verbrugge FH, Dupont M, Steels P, Grieten L, Swennen Q, et al. (2014) The kidney in congestive heart failure: ‘Are natri- uresis, sodium, and diuretics really the good, the bad and the ugly?’. Eur J Heart Fail 16:133-142.
- Schrier RW (1992) A unifying hypothesis of body fluid vol- ume regulation (The Lilly Lecture 1992). J R Coll Physicians Lond 26: 295-306.
- Schrier RW, Abraham WT (1999) Hormones and hemody-namics in heart failure. N Engl J Med 341: 577-585.
- Givertz MM (2001) Manipulation of the renin-angiotensin system. Circulation 104: e14-e18.
- Marenzi G, Lauri G, Assanelli E, Grazi M, Campodonico J, et al. (2002) Serum to urinary sodium concentration ratio is an estimate of plasma renin activity in congestive heart failure. Eur J Heart Fail 4: 597-603.
- Mentz RJ, Stevens SR, DeVore AD, Lala A, Vader JM, et al. (2015) Decongestion strategies and renin-angiotensin-aldo- sterone system activation in acute heart failure. JACC Heart Fail 3: 97-107.
- Nijst P, Verbrugge FH, Martens P, Bertrand PB, Dupont M, et al. (2017) Plasma renin activity in patients with heart failure and reduced ejection fraction on optimal medical therapy. J Renin Angiotensin Aldosterone Syst 18: 1-9.
- Hajime Kataoka (2019) Rational of the “chloride theory” as an explanation for neurohormonal activity in heart failure pathophysiology: Literature review. J Clin Exp Cardiolog 10: 634.
- Verbrugge FH, Nijst P, Dupont M, Penders J, Tang WHW, et al. (2014) Urinary composition during decongestive treatment in heart failure with reduced ejection fraction. Circ Heart Fail 7: 766-772.
- Testani JM, Hanberg JS, Cheng S, Rao V, Onyebeke C, et al. (2016) Rapid and highly accurate prediction of poor loop di- uretic natriuretic response in patients with heart failure. Circ Hear Fail 9: e002370.
- Ferreira JP, Girerd N, Medeiros PB, Santos M, Carvalho HC, et al. (2016) Spot urine sodium excretion as prognostic mark- er in acutely decompensated heart failure: The spironolactone effect. Clin Res Cardiol 105: 489-507.
- Doering A, Jenkins CA, Storrow AB, Lindenfelf J, Fermann GJ, et al. (2017) Markers of diuretic resistance in emergency department patients with acute heart failure. Int J Emerg Med 10: 17.
- Brinkley DM, Burpee LJ, Chaudhry SP, Smallwood JA, Lin- denfeld J, et al. (2018) Spot urine sodium as triage for effec- tive diuretic infusion in an ambulatory heart failure unit. J Card Fail 24: 349-354.
- Damman K, Ter Maaten JM, Coster JE, Krikken JA, Van Deursen VM, et al. (2020) Clinical importance of urinary sodium excretion in acute heart failure. Eur J Heart Fail 22: 1438-1447.
- Galluzzo A, Frea S, Boretto P, Pidello S, Volpe A, et al. (2020) Spot urinary sodium in acute decompensation of advanced heart failure and dilutional hyponatremia: Insights from DRAIN trial. Clin Res Card 109: 1251-1259.
- Hanberg JS, Rao V, Ter Maaten JM, Laur O, Brisco MA, et al. (2016) Hypochloremia and diuretic resistance in heart failure:Mechanistic insights. Circ Heart Fail 9: e003180.
- Nijst P, Verbrugge FH, Martens P, Dupont M, Tang WHW, et al. (2017) Renal response to intravascular volume expan- sion in euvolemic heart failure patients with reduced ejection fraction: Mechanistic insights and clinical implications. Int J Cardiol 243: 318-325.
- Honda S, Nagai T, Nishimura K, Nakai M, Honda Y, et al. (2018) Long-term prognostic significance of urinary sodium concentration in patients with acute heart failure. Int J Cardiol 254: 189-194.
- Martens P, Dupont M, Verbrugge FH, Damman K, Degryse N, et al. (2019) Urinary sodium profiling in chronic heart failure to detect development of acute decompensated heart failure. JACC Heart Fail 7: 404-414.
- Hajime Kataoka (2017) The “chloride theory”, a unifying hy- pothesis for renal handling and body fluid distribution in heart failure pathophysiology. Med Hypotheses 104: 170-173.
- Hajime Kataoka (2017) Proposal for heart failure progression based on the “chloride theory”: Worsening heart failure with increased vs. non-increased serum chloride concentration. ESC Heart Fail 4: 623-631.
- Hajime Kataoka (2015) Clinical significance of bilateral leg edema and added value of monitoring weight gain during fol- low-up of patients with established heart failure. ESC Heart Fail 2: 106-115.
- Kataoka H, Takada S (2000) The role of thoracic ultrasonog- raphy for evaluation of patients with decompensated chronic heart failure. J Am Coll Cardiol 35: 1638-1646.
- Hajime Kataoka (2007) Utility of thoracic sonography for followâ?up examination of chronic heart failure patients with previous decompensation. Clin Cardiol 30: 336-341.
- Espinel CH (1976) The FeNa test: Use in the differential diag- nosis of acute renal failure. JAMA 236: 579-581.
- Miller TR, Anderson RJ, Linas SL, Henrich WL, Berns AS,et al. (1978) Urinary diagnostic indices in acute renal failure.Ann Intern Med 89: 47-50.
- Harrison-Bernard LM (2009) The renal renin-angiotensin sys- tem. Adv Physiol Educ 33: 270-274.
- Castrop H, Lorenz JN, Hansen PB, Friis U, Mizel D, et al. (2005) Contribution of the basolateral isoform of the Na-K- 2Clâ?? cotransporter (NKCC1/BSC2) to renin secretion. Am J Physiol Renal Physiol 289: F1185-F1192.
- Holmer SR, Hengstenberg C, Mayer B, Engel S, Löwel H, et al. (2001) Marked suppression of renin level by β-receptor blocker in patients treated with standard heart failure therapy: A potential mechanism of benefit from β-blockade. J Intern Med 249: 167-172.
- Hajime Kataoka (2020) Proposal for new classification and practical use of diuretics according to their effects on the se- rum chloride concentration: Rationale based on the “chloride theory”. Cardiol Ther 9: 227-244.
- Schwartzenberg S, Redfield MM, From AM, Sorajja P, Nishimura RA, et al. (2012) Effects of vasodilation in heart failure with preserved or reduced ejection fraction: Implica- tions of distinct pathophysiologies on response to therapy. J Am Coll Cardiol 59: 442-451.
- Grodin JL, Simon J, Hachamovitch R, Wu Y, Jackson G, et al. (2015) Prognostic role of serum chloride levels in acute decompensated heart failure. J Am Coll Cardiol 66: 659-666.
- Grodin JL, Testani JM, Pandey A, Sambandam K, Drazner MH, et al. (2018) Perturbations in serum chloride homeostasis in heart failure with preserved ejection fraction: Insights from TOPCAT. Eur J Heart Fail 20: 1436-1443.
- Hajime Kataoka (2017) Vascular expansion during worsening of heart failure: Effects on clinical features and its determi- nants. Int J Cardiol 230: 556-561.
- Hajime Kataoka (2019) Biochemical determinants of changes in plasma volume after decongestion therapy for worsening heart failure. J Card Fail 25: 213-217.