
Advance in Environmental Waste Management & Recycling(AEWMR)
ISSN: 2641-1784 | DOI: 10.33140/AEWMR
Impact Factor: 0.9

Research Article - (2025) Volume 8, Issue 3
The Movement of Two Parallel Unpaired Electrons in Benzene and Aromatic Compounds
Received Date: Aug 01, 2025 / Accepted Date: Aug 29, 2025 / Published Date: Sep 04, 2025
Copyright: ©2025 Chun Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Citation: Wang, C. (2025). The Movement of Two Parallel Unpaired Electrons In Benzene and Aromatic Compounds. Adv Envi Man Rec, 8(3), 01-26.
Abstract
This year marks the 200th anniversary of Faraday's discovery of benzene, which is stable compared to other hydrocarbons. As we all know, Faraday made a huge contribution to electromagnetism. Coincidentally, benzene owes its special stability to the ring’s electric current and magnetic field. The oscillating hexagonal structure of benzene proposed by Kekulé 160 years ago is the most reasonable structural formula so far, which can be confirmed by modern infrared spectroscopy of the ring mode. However, the formation of the ring current and magnetic field has not been explained properly up to now. The problem lies in not having a correct understanding of the structure of the double bond.
Nuclear magnetic resonance has long confirmed that a double bond contains two unpaired electrons with parallel spins moving in two π-orbits separated by a covalent σ-bond. Due to thermal vibration, the σ-bonds are constantly stretched and compressed around their equilibrium position. The two π-orbits always revolve the two nearest carbon atoms, causing the π-orbits to shift and create an electric current. The current-induced magnetic field encompasses the benzene ring skeleton, making it stable. This additional magnetic field is a typical hallmark of aromatic stability and can be detected with 1 H-NMR spectroscopy. Various aromatic compounds are precisely associated with π-electrons including lone pairs. This paper will solve an aromatic conundrum that has puzzled the scientific community for more than a hundred years.
Keywords
Covalent σ-Bonds, π-Orbits, Lone Pair of Electrons, Aromaticity, Perimeter Ring Current and Magnetic Field
Introduction
In 1825, M. Faraday published an article "On New Compounds of Carbon and Hydrogen." This was the first discovery of benzene during the thermal decomposition of petroleum. He identified the substance as following [1]:
• Physical Properties: (The values in parentheses are the results of modern analysis). odor (sweet aromatic), colorless transparent liquid and specific gravity 0.85 at room temperature (density 0.876 g/cm3), crystalline specific gravity 0.956 in ice-cold water (1.012 g/cm3), melting point 42° F (41.95° F, 5.5° C). boiling point 176° F (176.2° F, 80.1° C), soluble in ether, alcohol, fixed and volatile oils. It does not conduct electricity.
• Chemical Properties: chlorine has little action on it until placed in sunlight. Iodine appears no action even in sunlight. Potassium exerts no action on the substance, even at a temperature of 186° F. Solutions of alkalis, or their carbonates has no action upon it. Nitric acid acts slowly upon the substance. Sulphuric acid exerts a moderate action upon it.
• Composition: hydrogen 1 and carbon 12 (in weight). Faraday believed that the substance contains 1 portion of hydrogen and 2 portion of carbon. At that time, the atomic weight of carbon was believed to be 6 atomic units. Therefore, he named the substance bi- carburet of hydrogen. Now we can understand that the composition of bi-carburet of hydrogen contains one portion hydrogen and one portion carbon. The 1:1 ratio of carbon to hydrogen and low chemical reactivity of benzene were a paradox to chemists in the early 1800's.
R. Kaiser published a paper “Bicarburet of Hydrogen”. Reappraisal of the Discovery of Benzene in 1825 with the Analytical Methods of 1968 [2]. The starting material used by Faraday must have been a very complex mixture of hydrocarbons, which would make severe demands even on present-day preparative analytical techniques, since it contains more than 300 components at concentrations greater than 100 ppm. Nevertheless, Faraday's persistence, experimental skill, and accuracy of measurement remain an example to the analyst of today.
In 1834, M. Mitscherlich published a paper “Ueber das Benzol und die Säuren der Oel- und Talgarten.“ [3]. By heating benzoic acid with lime, he obtained a substance having the physical and chemical properties completely the same as Faraday’s isolated benzene from oil gas. He determined its molecular formula C6H6, and the obtained hydrocarbon was named as benzin and later benzene, the name currently accepted by the IUPAC. In 1850, A. W. Hofmann recognized benzene and its derivatives as members of the great family of the so-called aromatic compounds [4].
These new aromatic compounds have attracted extensive attention from scientists around the world to reveal their chemical structures. Many structural formulas have been proposed historically. Among them, only Kekulé's benzene oscillation formula is the most reasonable one, although it is not perfect yet. In three articles, he gradually put forward his understanding of the structure of benzene. First, in 1865, he proposed in his article "On the Composition of Aromatic Substances" [5] that 6 carbon atoms form a six- membered closed chain, and 6 hydrogen atoms are connected to 6 carbons respectively. After that, a regular hexagonal structure was proposed in 1866.
He studied a lot of substitution products of the benzene and found the six hydrogen atoms on the closed ring are completely equivalent, then the six carbons must form regular hexagon in a plane arrangement [6]. In 1872, he pointed out that the formation of benzene from acetylene and the addition ability of benzene indicated the existence of alternating single and double bonds in the benzene ring. Biderivatives with positions 1.2 and 1.6 (C=C. and C-C) had to be necessarily different. But, no case of isomerism of the biderivatives of benzene (ortho-derivatives) was ever found. Kekulé stated that the atoms must be assumed in the systems, which we call molecules, in constant motion. The individual atoms of the system collide with each other in an essentially rectilinear motion in order to move away from each other again as elastic bodies.
In any case, the movement must be in accordance with the way that the atoms of the system are in the same relative arrangement, always to a middle equilibrium position to return. That is that the double bonds are in a state of constant oscillation such that any two adjacent carbon atoms were connected part of time by a single bond and part of time by a double bond. The idea of the movement of atoms in the molecule leaves us make themselves very nicely illustrated by phenakistoscopic images [7]. This oscillation model satisfies tetravalency of carbon. His benzene ring structure contains a dynamic change of double bonds, implying the formation of a ring current.
However, at the time, there was neither infrared (IR) spectroscopy, nor nuclear magnetic resonance (NMR) spectroscopy, and Kekulé had no experimental evidence to explain benzene’s stability. In contrast, Kekulé's oscillation structure should be a highly unsaturated and relatively unstable compound, completely contradicting the observed facts. Despite its imperfection, the oscillation model is just one step away from what we currently know about benzene structure 160 years later.
In 1929, K. Lonsdale published a paper “The structure of the benzene ring in C6(CH3)6” studied by using X-rays crystallography [8,9]. Since benzene itself is not crystalline at ordinary temperatures, the study of the benzene nucleus or ring has had to be referred to certain of its derivatives, such as C6(CH3)6. The benzene nucleus is a planar ring of six carbon atoms with a diameter of 1.42 Å, such as occurs in the graphite structure. The ring itself and all the sidechain carbon atoms lie in one plane. Lonsdale confirmed the planar, cyclic, equal bond length of the Kekulé’s benzene structure.
In 1930 and 1931, H. Mark and R. Wierl [10,11] used electron diffraction to study the atomic arrangement of benzene vapor. This is the interference image explained when C atoms are arranged in a regular hexagon and the distance between adjacent C atoms is assumed to be 1.4 Å.
In 1976, K. Tamagawa, T. Lijima and M. Kimura determined the molecular structure of benzene by combining the average distances obtained by the present electron diffraction study and the moments of inertia. The thermal average bond distances have been determined: (C-C) = 1.399 ± 0.001 Å and (C-H) = 1.101 ± 0.005 Å [12].
In 1931, E. Hückel pointed out that “The particular behavior of benzene (and other "aromatic" substances compounds), its stability and its "aromatic" character, in contrast to other compounds with double bonds, can therefore be even then not expressed with the help of Kekulé's formulation”. The number of ring atoms does not have to be 6, for example, the aromatic five-member ring furan and thiophen. The cyclopentadiene seems to have the same structure as furan and thiophene with five-member ring and two double bonds, but it is not aromatic. However, cyclopentadienyl potassium is aromatic. The formulation of rings with single and double bonds (also if this is assumed to be "flowing" in the sense of the oscillation hypothesis) cannot explain the aromaticity of cyclopentadienyl potassium etc. To explain the stability of the aromatic ring and the equalized bond length, Hückel [13-15] introduced the newly proposed quantum mechanics.
The electrons outside the nucleus of each atom are divided into two categories: the paired electrons in the inner shell and the paired electrons of the covalent bond do not participate in aromatic stability, and the configuration of the remaining unpaired electrons is the source of aromaticity. There are six unpaired electrons in the benzene ring that are in the six p-orbitals of quantum mechanics and they are perpendicular to the plane of the benzene molecule. The overlap of the p- orbitals is the cause of the ring current. Furan, etc., all have six unpaired electrons, so they are the same as benzene and have aromatic properties. However, Hückel didn’t give the configuration of the electrons on the heteroatoms, such as oxygen in furan.
Hückel used F. Bloch's theory of electron delocalization in metal lattices [16]. This theory itself has a fatal error. Electrons do not swim in the lattice, but revolve around atomic nucleus at high speed. He also introduced the E. Schrödinger function [17]. Schrödinger's theory itself was a mistake. It is impossible for electrons to appear according to probability. Hückel described the states of n electrons in an atomic field of N atoms. Looking at his matrix calculations, one can immediately see that his theory is very unlikely, because the benzene ring has only six carbons and six hydrogens, which is not so complicated. He mentioned that with the interpretation of the electron configuration given here benzene loses the Kekulé’s formulation with alternating double and single bonds, it makes sense.
His core content of benzene molecular orbital is the delocalization of electrons. The stability, aromaticity, and equalized bond length are generally attributed to the two delocalized circles above and below the ring. With this model, the ring current detected by 1H-NMR can be explained well. Hückel’s empirical rule is very useful to predict the aromaticity of compounds. 1. Cyclic, 2. Planar, 3. Alternating single and double bonds, 4. 4n+2 active electrons (unpaired electrons), (n=1, 2, 3, … natural numbers).
In 1933, L. Pauling and G. W. Wheland presented the quantum- mechanical calculation of the resonance energy of benzene [18]. He gave five canonical structures contributing to the normal state of benzene molecule. The principal contributions to the structure are made by the two Kekulé structures, resonance between them stabilizing the molecule to the extent of 0.9α over a ring with three double bonds.
It should be said that Hückel proposed the delocalization of electrons on the benzene ring and Pauling, on the other hand, proposed a valence bond theory for benzene ring.
In 1986, D. L. Cooper et al. published an article in Nature questioning the delocalization of electrons in the benzene ring [19]. They stated that the electrons in benzene were almost certainly localized to particular carbon atoms and the aromaticity of benzene came from spin-orbit coupling rather than electron delocalization. This is the first paper to suggest the existence of electron spins and orbits in benzene rings, rather than spin degeneracy mentioned by Pauling. However, the additional magnetic field caused by the ring current has been confirmed by NMR, this fact cannot be explained by the localized spin-orbit coupling.
In 1987, J. Maddox published a paper “Benzene’s electron structure” in Nature [20]. He stated “A new calculation shows that Kekulé may have made a good model of benzene, but also fails to resolve the question whether valence bonds are better than molecular orbitals (or vice versa)”. The new calculation was performed by P. A. Schulz and R. P. Messmer and published in the paper "Are π bonds present in benzene? [21]. They pointed out that the bent-bond description is energetically the better representation of the ground state. Their particular contribution was their concept of what they call the "bent-Q" bond. An electron has the same charge value as a proton, but its mass is 1/1800 of the proton’s mass. The electrons must revolve around the positive charges in the molecule at extremely high speed, otherwise they will be sucked into the nucleus. This speed is about 106 m/s, which means that the electron can circle the earth three times in one minute. Therefore, electrons cannot move in bent-Ω bonds, or there are absolutely no bent bonds.
In 2009 atomic force microscopy (AFM) was used to make the atoms in benzene rings (pentacene) visible for the first time [22]. However, there is still a gap between the atomic arrangement and the electron movement. Being able to see the benzene ring structure cannot explain the magnetic field and aromatic stability caused by its ring current.
Up to now the benzene's structure continued to puzzle the world's greatest scientists. In 2020, Yu Liu et al. published an article: “The electronic structure of benzene from a tiling of the correlated 126-dimensional wavefunction” [23]. It is reportly that after 90 years, scientists reveal the structure of benzene. One of the fundamental mysteries of chemistry has been solved by a collaboration between Exciton Science, UNSW and CSIRO – and the result may have implications for future designs of solar cells, organic light-emitting diodes and other next gen technologies.
Such a simple C6H6 has been studied more and more complexly. According to report, Bohr once told Pauling that to study the structure of the benzene ring, one must understand the structure of the double bond. This is the key issue. By adding the correct double bond structure to the Kekulé’s structure, all aromatic compounds can be explained consistently.
The first part of this work introduces the electron motion in single and double bonds, and briefly explains the principle of NMR in an easy-to-understand manner. The second part introduces the Kekulé oscillation formula of benzene ring and the phenakistoscopic images. NMR results confirmed the coexistence of the double bond magnetic field and the ring current induced magnetic field. Increasing temperature, the aromatic ring turns into a double- bonded ring, which is strong evidence to prove the correctness of the Kekulé’s oscillation structure. Section 3 introduces the Hückel’s 4n+2 rule and discusses the stability of polycyclic aromatic hydrocarbons (PAH).
In section 4, lone pair of electrons is introduced, which can be treated as small π-electrons to interpret the heterocyclic aromatic compounds. Section 5 discusses the aromaticity of charged ring system. The formation of the ring current can be the repulsive force between the negative charges (π-electrons and lone pair of electrons) or the attraction of the positive charge (C+) to the negative charges, as long as the number of active electrons conforms to Hückel’s 4n+2 rule. Section 6 answers why molecules of cyclic, planar, 4n+2 are aromatic and 4n are antiaromatic. In the last section 7, two special large cyclic aromatic compounds (C18H6 and C18) containing triple bonds and cumulene double bonds are discussed. The shift of the π-orbits by thermal vibration is the key for aromaticity.
All experimental facts proved the correctness of Kekulé's idea of aromatic ring oscillation, but the Kekulé model lacked a description of magnetism.
Methodology
This work is mainly based on the experimental data of nuclear magnetic resonance, infrared spectroscopy and X rays-diffraction, combined with the oscillation model proposed by Kekulé. The electron motion under various situations was drawn by using Power Point Presentation.
The author has been working in the fields of chemistry, physics and materials science for many years and is familiar with various experimental instruments. Due to the comprehensive integration of knowledge, the author was able to develop a reasonable understanding of some of the confusing concepts presented in currently published scientific articles. The logical chain in the reasoning process is based on experimental facts provides by many previous scientists.
Results & Discussion
Covalent Single σ-Bond and Conjugated Double Bond
The covalent bond (σ-bond) is the most important bonding in nature. It involves the sharing of two electrons between two adjacent atoms. But how to share?
An electron is a negatively charged particle, which had already been determined by J. J. Thomson (Nobel Prize in Physics, 1906) and R. A. Millikan (Nobel Prize in Physics, 1923) as early as 1897 and 1909 respectively [24, 25]. Electrons can be attracted by any positive electric field. When two atoms get closer, the higher- energy valence electron (chemical name of unpaired electron) in one atom can also be attracted by the other nucleus, and vice versa.
The maximum superimposed electric field of two identical nuclei is in the fixed ring, as shown in Figure 1a. Once the Coulomb attractive forces between the nuclei and the electrons, the repulsive force between nucleus-1 and nucleus-2, as well as the centrifugal forces of the circling electrons are balanced, the two unpaired electrons from the two atoms enter the fixed ring region with antiparallel spins, which cancel out the original orbital magnetism and lower the system energy. After that, the two electrons travel in the new orbit and form a new covalent bond. This should be a reasonable structure of a covalent bond.
So far, descriptions of covalent bonds have not considered the factors such as the charge and mass of electrons and the charge and mass of nuclei.
The Lewis dot structure is far from reality. Maximum overlap of orbitals introduces a very wrong concept. “A molecular orbital is a mathematical function describing the location and wave-like behavior of an electron in a molecule. This function can be used to calculate chemical and physical properties such as the probability of finding an electron in any specific region.” In fact, the positive electric fields determine how electrons move. Electrons cannot form electron clouds and be discovered by probability.

Figure 1: The Formation of a Two-Electrons Covalent σ-Bond (a) and a One-Electron Covalent σ-Bond (b). The Electrons Circle in The Maximum Superimposed Electric Field of Two Positively Charged Carbons.
In 2024, T. Shimajiri et al. published a paper in Nature “Direct evidence for a carbon–carbon one-electron σ-bond” [26]. Raman spectrum verified the presence of a C•C one-electron σ-bond by C-C symmetric vibration at 379 cm-1 peak. Single-crystal X-ray diffraction analysis confirmed the elongated C-C single bond of 2.92 Å at 100K. This experimental fact can be fully understood by using the electronic structure of the covalent bond in Figure 1a. The C1+ and C2+ superimposed electric field can enter a single electron or enter two electrons with antiparallel spins. Of course, if there is only one negatively charged electron rotating and sharing between C1+ and C2+, the positive electric repulsion between C1+ and C2+ will be greater than in two-electrons σ-bond. Therefore, the one-electron σ-bond is 2.92 Å as shown in Figure 1b. The two- electron σ-bond is about 1.54 Å with Raman shift at 1155 cm- 1.Comparing the two σ-bonds, it can be seen that one-electron σ-bond (ca. 0.047 eV) is much weaker than typical two-electrons σ-bond (ca. 1.43 eV). It is absolutely impossible to explain single- electron covalent bond in terms of maximum overlap of orbitals.
As early as 1931, Pauling proposed a single-electron covalent bond based on the existence of H •+ radical cation. The single electron didn’t attach to H1 or H2, but shared between them. Only when the unperturbed system is degenerate or nearly degenerate, as in H + where the two nuclei have the same charge, does there exist a resonance energy leading to molecule formation [27]. His explanation is not as clear and accurate as Figure 1b. Single- electron covalent bonds can occur between nuclei with different charges. It is the superimposed electric field, not the resonance energy, that determines the formation of covalent bonds. When the temperature drops to absolute zero, the resonance energy tends to zero, but the covalent bond does not disappear.
Double bond has always been a mystery in organic chemistry. It is not twice as strong as a single bond. On the contrary, the chemical reactivity of a double bond is much higher than that of a single bond. The most reasonable model to date shows that a double bond is constructed by a covalent σ-bond and a conjugated π-bond. However, various descriptions of conjugated π-bond, such as the overlapping of two p-orbitals and the delocalization of electrons in large π-bond, still cannot explain the fact that double bonds have color activity, electrical conductivity, chemical reactivity, and ring aromaticity.
The most active properties of matter should be traced back to unpaired electrons. Surprisingly, the graph (Fig. 2a) explaining the 1H-NMR result in organic chemistry textbook [28] precisely implies that there are two parallel unpaired electrons in the double bond. It was stated in the textbook that the whirl-shaped magnetic field around the double bond was induced from the electron cloud by an external magnetic field. What is an electron cloud? The electron cloud is the probability of finding any electrons in any specific region around the nucleus. How does the electron cloud become two elliptical orbits under the action of an external magnetic field? Obviously, the cloud model based on the Schrödinger theory is very vague.

Figure 2: (a). 1H- NMR Confirmed Double Bond Structure. (b). New Proposal of Double Bond. Two Parallel Unpaired Electrons Describe Two π-Orbits Above and Below The σ-Bond. The Two Unpaired Electrons Make the Double Bond Magnetic.
An electron is a self-rotating charged tiny magnet. In an external magnetic field, the electron spin (discovered by G. Uhlenbeck and S. Goudsmit in 1925 [29]) aligns with the magnetic field lines. The electron rolls around the nucleus and its spin is perpendicular to its orbit, which is the lowest energy state.
Based on the prompt of the graph of Figure 2a, a new structure of the double bond is proposed, as shown in Figure 2b. The covalent σ-bond is established by sharing two electrons from the sp2 hybrid orbits of two adjacent carbon atoms. The conjugated π-bond is built up by two completely same unpaired electrons with parallel spins. These two electrons are separated by the σ-bond. Each π-electron travels around two nuclei and describes an elliptical orbit. These two unpaired electrons should be the origin of the induced whirl- shaped magnetic field in the external magnetic field. In order to avoid confusion, π-bond will be changed to π-orbits or π-electrons, because these two parallel unpaired electrons are not bonded to atoms, but can flow. This is the key to understanding aromatic ring currents.
NMR technology can identify aromatic compounds. It is necessary to briefly introduce the basic principle of 1H-NMR. In an external magnetic field (B ), the 1H spins strive to be oriented with the magnetic field lines. However, a proton is 1800 times heavier than an electron, so this orientation cannot be fully developed, and it is still accompanied by procession motion, just like a spinning top. The stronger the external magnetic field, the greater the orientation, the higher the energy required to flip-flop the proton spin from the α-state to the β-state, as shown in Figure 3
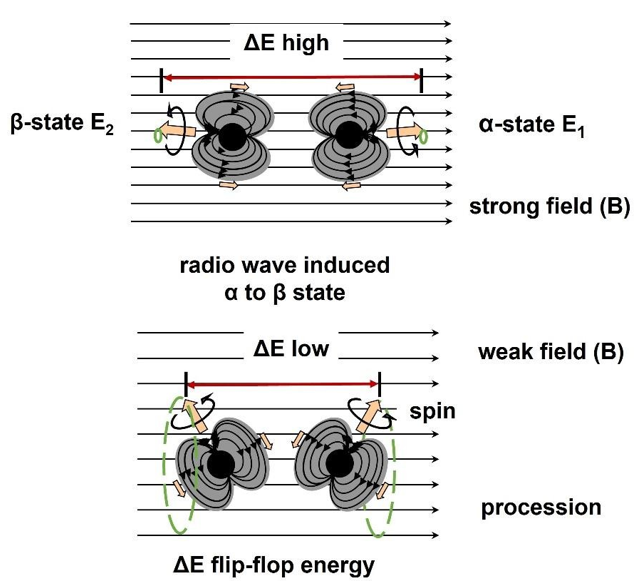
Figure 3: Basic Principle of 1H-NMR. The Denser the Magnetic Field Lines, The Higher the Flip-Flop Resonance Energy of The Proton.
Using radio waves to impact proton, if the frequency of the wave exactly matches the energy of the spin direction reversal, the spin will turn over to the higher energy β-state. Switching off the radio wave, the proton returns to the lower energy α-state and releases a peak. In the experiment, the 1H-NMR peak of tetramethylsilane (TMS) is used as a reference. This molecule has only one kind of proton spins, and other atoms are not magnetic, so its 1H-NMR spectrum consists of a singlet. The resonant frequency (energy) of the TMS is ν0, and the resonant frequency of the study sample is ν. The chemical shift is defined as following:
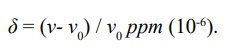
When certain groups in the molecule are magnetic, if the local magnetic field at the 1H position is strengthened, the resonant energy increases, so the chemical shift is positive (deshielded); if the local magnetic field is weakened, the resonant energy decreases, so the chemical shift is negative (shielded).
Kekulé Oscillation Structure of Benzene Ring
Kekulé refined his understanding of the structure of benzene in three articles [5-7] in 1865, 1866 and 1972 respectively. Benzene has a closed-ring structure, regular hexagonal shape, with three double bonds in the ring, and has a certain ability for addition reactions. “Due to the oscillation motion of the carbon atoms (thermal vibration), any two adjacent carbon atoms were connected part of time by a single bond and part of time by a double bond (Fig. 4). The idea of the movement of atoms in the molecule leaves us make themselves very nicely illustrated by phenakistoscopic images” (Kekulé 1872). A snapshot picture of this dynamic image is shown in Figure 5. However, he was not able to explain the aromatic stability of benzene

Figure 4: The Original Kekulé’s Oscillation Structure of Benzene Ring. Any. Two Adjacent Carbon Atoms Were Connected Part of Time by A Single Bond and Part of Time by A Double Bond
The infrared spectrum of benzene shows the carbon-carbon stretching and compressing vibration in the range of 1600-1400 cm-1, which is a so-called ring mode. Converted the wave number to wavelength, it is λ=6.25-7.14 µm. Based on λ=c/ν, (c, speed of light, ν, frequency), the vibration frequency is about 1014 Hz, namely, the C-C bond in benzene ring vibrates 1014 times per second. An electron must revolve in a molecule at ultra-high speed of 106 m/s, otherwise, it would be attracted into the carbon nuclei.
This speed means that an electron can travel around the earth three times in a minute! The bond length of benzene is about 1.40 Å. The π-electrons first revolve around two carbon atoms about many times before they meet next vibration motion of the σ-bond and change their position, because the π-orbits prefer to revolve around two carbon atoms with shortest distance. The 1014 Hz vibration makes the single and double bonds to look like equalized. The imaging microscopy technology is still unable to capture the molecular structure that appears in picosecond (10-12 s). Therefore, single and double bonds cannot be distinguished in the benzene molecule, but they do exist. If the π-orbits move in one direction, (i.e., the phenakistoscopic image in Figure 5 mentioned by Kekulé), an electric current is then formed and a magnetic field is induced around the benzene skeleton. Just the ring current and the additional magnetic field are the key to aromatic stability.

Figure 5: A Snapshot Picture of The Phenakistoscopic Images Mentioned by Kekulé in 1872. The Double Bonds in The Benzene Ring Shift Counterclockwise Along the Perimeter to Form Ring Current.
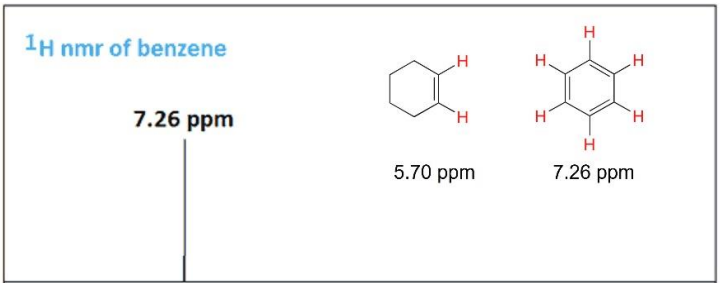
Figure 6: Comparing the 1H-NMR Spectra of Cyclohexene and Benzene. The Chemical Shifts of the Hydrogens Attached to The Double Bonds are 5.70 ppm and 7.26 ppm, Respectively.
Faraday and Kekulé, both of them mentioned that under the same conditions, addition reactions do not easily occur on the benzene ring, while can occur on cycloalkenes, so the benzene ring has a certain stability, as shown in Figure 7.

Figure 7: Direct Evidence of the Aromatic stability of Benzene Ring. Substitution Reaction (Benzene) Instead of Addition Reaction (cyclohexene) on the Double Bonds.
The newly proposed double bond structure introduced into Kekulé’s oscillation structure, can make the benzene structure perfect, as shown in Figure 8. During thermal vibration, the σ-bonds stretch and compress, leading to the shift of π orbits. “Any two adjacent carbon atoms were connected part of time by a single bond and part of time by a double bond” (Kekulé1872).
Different from the currently popular electron delocalization, the magnetic field around benzene molecules comes from two parts: the ring current and the π-orbits, as shown in Figure 9. A convincing experimental fact supporting the existence of double bonds in aromatic compounds is the 1H- NMR result of [14] annulene. At -60°C, two signals, one at 0 ppm and one at 7.6 ppm, appeared in the NMR spectrum [30], as shown in Figure 10a. If only the ring current induces a magnetic field, the outside of the ring is paramagnetic and the inside is diamagnetic. The inner 4 protons should experience negative chemical shift δ<0. However, δ=0 ppm means that there must be a paramagnetic field inside the ring balancing the diamagnetic field. The π-orbits induced magnetic field inside and outside the ring are both paramagnetic, which balances the ring’s diamagnetic field, as shown in Figure 10b. Coincidentally, the chemical shift of the protons in the [14] annulene ring is zero, revealing the indisputable fact that double bonds still exist in aromatic compounds.

Figure 8: During the Expansion and Compression of The Thermal Vibrations Of the σ-Bond, the π-Orbits Shift, Forming the Resonant Structure of Benzene. The Vibration Frequency Is About 1014 Hz, Which Makes the Bond Lengths Equal.

Figure 9: (a). The Constant Movement of the π-Orbits Along the Perimeter of the Benzene Generates Ring Current, and the Associated Ring Magnetic Field. (b). The Magnetic Fields of the π-Orbits Remain in The Molecule.
The ring current is formed by the jumping motion of the π-orbits, just like the formation of electric current in metals by the jumping motion of the unpaired electron orbits. The higher the temperature, the larger the thermal vibration amplitude and the greater the distance between the atoms. When the π-orbits can no longer jump over any more, there will be no ring current and the double bonds are fixed. The aromatic [18] annulene is an excellent example illustrating this behavior. “1H-NMR spectra of [18] annulene are temperature-dependent and show a singlet at 5.45 ppm at 120°C and two multiplets at 9.25 ppm (12H) and −2.9 ppm (6H) at −60°C [31]”. The 12 outer protons experience chemical shift at 9.25 ppm, which is typical ring current signal (Fig. 11a). However, at 120°C, the same molecule shows a single chemical shift at 5.45 ppm, which is a typical cycloalkene double bond signal (Fig. 11b).

Figure 10: (a). The Aromatic [14] Annulene Shows the 1H-NMR Shifts Inside (4H) 0.0 ppm, Outside (10H) 7.6 ppm. (b). Ring Current Induced Magnetic Field, Outside Is Paramagnetic, Which Strengthen the External Magnetic Field (B0), Inside Is Diamagnetic, Which Weakens the B0. The π-Orbits Induced Magnetic Field Inside and Outside the Ring Are Both Paramagnetic, Which Balances the Ring’s Diamagnetic Field.

Figure 11: (a). At -60°C, [18] Annulene Shows Typical Aromatic Chemical Shifts (12H 9.25 ppm, 6H -2.9 ppm) of 1H-NMR. (b). At 120°C, Typical Cycloalkene Chemical Shift (18H 5.45 ppm). The Higher the Temperature, the Greater the Amplitude of Thermal Vibration, and the Farther Apart Carbon-Carbon Is. If The π-Orbits Cannot Jump Over Any More, There Will Be No Ring Current and The Double Bonds Are Fixed.
In fact, [14] annulene also shows the temperature-dependent ring current phenomenon. The chemical shift of 7.6 ppm caused by the ring current at -60°C disappears at room temperature.
Another paper reported that the ring current disappears, when replaces a member with longer bond length. The ring current is not formed by delocalization of electrons, but by constantly changing positions of the double bonds. These experimental facts confirmed the Kekulé oscillation benzene formula.
Polycyclic Aromatic Hydrocarbon (PAH)
Hückel’s rule
The compounds must meet the following four conditions to generate ring current and consequent aromatic stability.
1) Cyclic
2) Planar
3) Alternating single and double bonds
4) Containing 4n+2 active electrons (n integer).
The first three conditions are easy to understand and will not be discussed in detail.
What are active electrons?
Active electrons should be unpaired electrons. The ring current is always related to the double bond, which has two unpaired electrons in two π-orbits separated by a σ-bond. When n=1, then 4n+2=6, 6/2=3. This is the number of double bonds in the benzene ring. When n= 2, 3, 4, and then (4n+2)/2=5, 7, 9, etc. They are odd numbers. The Hückel’s rule should be that the molecule contains an odd number of double bonds.
Pyrene has a cyclic, planar, alternating double bond and single bond structure, but it contains 8 double bonds (an even number) and does not obey Huckel's rule. How to understand its aromatic stability?

Figure 12: Three Expressions of Pyrene: (a), (b) and (c). The Addition Reaction Occurs Only at the Central Double Bond (d). Expression (c) is Correct, Because the Ring Current and Magnetic Field Can Stabilize the Double Bonds Along the Perimeter of The Molecule.
The molecular structure of pyrene has three expressions (a), (b) and (c), as shown in Figure 12. It is generally believed that its aromaticity is due to the four benzene-like rings (b). When another organic compound was added to the system, the addition reaction occurred only at the central double bond (d). The other seven double bonds remained unchanged. This means that formula (c) is correct. The experimental evidence indicates that seven double bonds along the perimeter of the molecule participate in the ring current, which obeys the Hückel’s rule. The 1H-NMR results confirmed the ring current along the perimeter. All protons are in the deshielded magnetic field outside the ring. Since the environmental differences are not large, the chemical shifts are 7.987 ppm, 8.161 ppm and 8.054 ppm respectively (7.26 ppm benzene, 5.70 ppm cyclohexene). The ring current and magnetic field stabilize the seven double bonds so that no addition reactions occur.
The next two examples (anthracene and phenanthrene) show that the molecules with large ring currents around their perimeters require additional energy to convert to their mesomeres. This means that the former is more stable than the latter, as shown in Figure 13. Therefore, the position of double bonds in a molecular structure cannot be arbitrarily labeled.

Figure 13: Anthracene and Phenanthrene With Large Ring Currents Along Their Perimeters Require Additional Energy to Convert intoTheir Structural Isomers. The Formers Are More Stable Than The Latter’s.
Many polycyclic aromatic hydrocarbons (PAH) have been described more than one chemical formula. The following three examples, picene, naphthalene, and coronene, have at least two structural isomers, which can often be seen in organic chemistry textbooks and in the internet (Fig.14).

Figure 14: Correct Expressions of The Polycyclic Aromatic Hydrocarbons (PAH). The Top Row Is Wrong and The Bottom Row Is Right. The Position of The Double Bonds Cannot Be Arbitrarily Marked on The Molecular Structure.
In terms of the molecular stability, the expressions for the bottom row are correct because they can produce larger perimeter ring currents.
Heterocyclic Aromatic Compounds and Lone Pair of Electrons
Heterocyclic aromatic compounds refer to compounds in which one or more carbon atoms are replaced by other atoms in the carbocyclic framework, and have aromaticity.

Figure 15: Four heterocyclic aromatic compounds. The Two Dots Inside the Ring Are Lone Pair of Electrons That Participate in The Ring Current, While the Two Dots Outside the Ring Are Lone Pair of Electrons That Do Not Participate in The Ring Current.
The four molecules in Figure 15 are exactly what Hückel presented in his paper “Quantum theoretical contributions to the benzene problem” in 1931 [14]. These four compounds, like benzene, exhibit the aromatic character of a series of substitution reactions. However, alternating single and double bonds are not present in the last three compounds. Kekulé’s oscillation structure was questioned. This was one of the problems with benzene. Hückel found the four heterocyclic aromatic compounds all have 6 unpaired electrons in the ring, which is the key for aromatic stability. Benzene can be described by overlapping of six p-orbitals, but pyrrole, furan and thiophene cannot be described by the six p-orbitals. He introduced Bloch’s hypothesis that electrons can swim in metallic crystal lattice. Therefore, the six electrons in the five-member ring are simply delocalized. He thought it makes sense to lose the alternating single and double bonds in this way. This was the beginning of the electron delocalization hypothesis that is still influential today. However, in Section 2 we clearly saw that double bonds are still present in aromatic compounds. The electron delocalization hypothesis had to be questioned.

Figure 16: (a). Furan has two lone pairs of electrons on the oxygen site, similar to a water molecule (b). The Resulting Molecule Is Not Planar and Does Not Match the Aromatic Stability of Furan.
Now we take furan as an example to analyze the generation of ring current and aromatic stability. According to known knowledge, the six outermost electrons of the oxygen atom are sp3 hybridized (Fig. 16a), similar to the oxygen in water molecule (Fig. 16b). Two of the electrons are covalently bonded to two carbon atoms. The remaining four electrons enter two elliptical orbits as two lone pairs. However, the resulting molecule is not planar, and does not match the aromatic stability of furan. These two lone pairs must be reorganized. As shown in Figure 17, one lone pair of electrons composed of two parallel unpaired electrons above and below the molecular plane revolve around the oxygen atom, just like π-electrons, participating in the ring current. The other lone pair of electrons travels in an elliptical orbit coplanar with the molecular plane and does not participate in the ring current. As long as the lone pairs of electrons are treated as small π- electrons, the structure satisfies the four conditions for aromatic compounds.

Figure 17: After Reorganization, The Lone Pair Participating Ring Current, Acts as Small π-Electrons to Form 6π-electrons in Furan, Which Satisfy Hückel’s Rule (4n+2). The Unpaired Electrons in the π-Orbits Will Repel the Lone Pair in Front, Forcing It to Move Forward. The Other Lone Pair of Electrons Travels in An Oval Orbit Coplanar with The Molecular Plane and Do Not Participate in The Ring Current. The 1H-NMR Chemical Shifts Are 6.380 ppm and 7.435 ppm, Indicating the Existence of Ring Current.
Once the π-orbits (Nr.1) change position due to the stretching and compressing vibration, the unpaired electrons in the π-orbits will repel the lone pair of electrons (Nr.2), forcing them to move forward. Further changing position of the π-orbits (Nr.3), leaves a charge-deficient carbon atom. The lone pair of electrons (Nr.2) will then revolve around two carbon atoms, and the π-orbits (Nr.1) will revolve around the oxygen atom as a lone pair. When unpaired electron orbits (π-orbits and lone pair) flow along the molecular skeleton, a ring current is generated, which induces a ring magnetic field, resulting in aromatic stability. The 1H-NMR signals at 6.380 ppm and 7.435 ppm proved the existence of ring current in furan. Thiophene and pyrrole should have an aromatic five-membered ring structure similar to furan. Pyridine is similar to a benzene ring.
Lone pair of electrons is not sufficiently understood. Usually only two Lewis dots are drawn on the orbit. Since it is called an electron pair, both electrons must be present at the same time. What force holds the negatively charged electrons together? Analogous to the π-electrons, the whirl-shaped magnetic field enveloped the orbits of two parallel unpaired electrons, as shown in Figure 18. Unlike π-electrons, lone pair has 360° rotation freedom along its long axis of the ellipsoid. Like π-electrons, lone pair has certain physical activity and certain chemical reactivity and is magnetic.

Figure 18: The Configuration of Lone Pair of Electrons has 360° of Freedom About the Long Axis of The Ellipsoid. Lone Pair Has Magnetic Property.
The magnetism of lone pair of electrons is rarely mentioned, but this property plays an important role in materials. Water drop (5 cm) can be levitated in strong magnetic field, showing strong diamagnetic property. The O-H covalent bond in water molecule is not magnetic, and the oxygen atomic core is not magnetic. Protons exert only a weak paramagnetic effect on their surroundings. Therefore, the strong diamagnetism of water drop comes exclusively from two lone pairs of electrons. In the absence of an external magnetic field, the lone pair has 360 degrees of rotation-freedom, therefore, it does not show magnetic properties. Applying external magnetic field, the electron spins orient in the magnetic field lines, and then the lone pair turns from ellipsoid into two elliptical lines. Water is a condensed matter and its lone-pair- orbits are densely packed; the system shows diamagnetism.
Although the lone pair of electrons outside the aromatic ring does not participate in the ring current, its magnetic field will affect the 1H-NMR signal of aromatic compounds. For example, in pyridine, the magnetic field of the lone pair of electrons outside the ring is superimposed on the magnetic field induced by the ring current, causing the protons of the C(A)-site to experience a large chemical shift at 8.613 ppm. However, the pyrrole at the same C(A)-site shows chemical shift of 6.737 ppm, which is much lower than that of pyridine, because there is no lone pair outside the pyrrole ring, as shown in Figure 19.

Figure 19: The Magnetic Field of The Lone Pair Out of Ring Makes Extra Contribution to The Local Magnetic Field of The Ring Current. The Ortho-H in Pyridine Has 1H-NMR Chemical Shift at 8.613 ppm, While Ortho-H in Pyrrole at 6.737 ppm.
Aromaticity of Charged Ring System
There is a striking relationship between the stability of the charged ring systems, both cationic and anionic, and their aromaticity (or antiaromaticity), as shown in Figure 20 [32]. In this section, the generation of ring currents for the first four aromatic charged species will be discussed.

Figure 20: Overview of Some Aromatic and Antiaromatic Charged Rings.
Cyclopentadienyl anion (Cp-) is an aromatic five-membered ring, which has comparable 1H-NMR chemical shift to the isoelectronic (but uncharged) benzene [33]. In 2016, S. Bachmann et al. reported the 1H-NMR results of alkali-metal cyclopentadienides. For example, Cp-Li showed the 5H chemical shift at δ=5.69 ppm and the Li chemical shift at δ= -7.75 ppm [34]. Lithium is at the center of the five-membered ring and above the ring plane. This is clear evidence that Cp- has aromatic ring current. Inside is diamagnetic and outside is paramagnetic. How to understand the formation of this aromatic anion?
Figure 21 presented the formation of the Cp-. The precursor cyclopentadiene undergoes sp3 hybridization at the carbon-1 position. Two hydrogen atoms are attached to this carbon. This molecule is non-planar and has no aromaticity. If one of H-C(1) covalent bond is broken, H and C(1) will respectively withdraw their shared electrons. There is an unpaired electron at C(1) position. Although the molecule at this step is electrically neutral, but it is an unstable radical. When H+ leaves the molecule and gives the C(1) another electron forming an anion. The two unpaired electrons at C(1) do not combine with other atoms and at the same time, C(1) hybridizes from sp3 (3D) to sp2 (2D) to reduce the system energy. Because of the participating in the ring current, the two unpaired electrons change into lone pair. Now that the number of active electrons satisfies the Hückel‘s rule (4n+2), or 6 π-electrons the aromaticity of the cyclopentadienyl anion can be understood. The resonance structure is exactly the same as furan, pyrrole and thiophene.

Figure 21: The Aromatic Cyclopentadienyl Anion (Cp-) is Formed from The Precursor Cyclopentadiene. Lone Pair Participates in Ring Current. This System Has 6π Electrons Similar to Furans. Cp-Li Shows a 5H Chemical Shift of δ=5.69 ppm and a Li Chemical shift of δ= -7.75 ppm.
In 2004, T. Matsuo and A. Sekiguchi published a paper “Cyclobutadiene Dianion” [35]. The dilithium salt of the cyclobutadiene dianion has aromatic properties, as shown in Figure 22. Although the parent cyclobutadiene dianion is not easily obtained, its derivatives, for example [Li+ ]•[(Me Si) C 2-] current in cyclobutadiene dianion has not yet been satisfactorily explained.
One of the two double bonds of cyclobutadiene is opened (Fig. 22d). Two extra electrons combine with the opening double bond 2 3 4 4 (Fig. 22a) is stable. The four membered ring of carbons is planar, square, with equal bond lengths. The two Li+ ions are located above and below the center of the plane of the four-membered ring (Fig. 22b). These were determined by X-ray crystallography. The 6Li-NMR showed a singlet chemical shift at -5.07 ppm. This indicates that there is ring current in the four-membered ring. The induced whirl-shaped magnetic field is paramagnetic outside the ring and diamagnetic inside the ring. The Li ions are located in the weakened magnetic field (Fig. 22c). The mechanism of the ring to form two lone pairs of electrons with parallel spins (Fig. 22e). In this way, there are six π-electrons in the four-membered ring. Under thermal vibration, the lone pairs and the original π-electrons constantly change position at 1014 Hz due to electron-electron repulsion. As a result, the ring current is formed associated with magnetic field enveloped the ring skeleton and the bond lengths are equalized. The same mechanism as furan and cyclopentadienyl anion.

Figure 22: (a). Aromatic cyclobutadiene dianion. (b). Two Li+ ions are located above and below The Center of The Plane of The Four- Membered Ring. (c). The Ring Current Induced Magnetic Field Inside the Ring Is Diamagnetic (Li, δ=-5.07 ppm). (d). One Double Bond Is Opened to Form Two Radicals. (e). Two Extra Electrons Enter the System to Combine with The Two Radicals to Build Two Lone Pairs, to Form 6π-Electrons System.
The cyclopropenyl cation (CPCs, cyclopropenium ion, C3 H3 +) has been found to possess extraordinary stability despite being both cationic and highly strained (Fig. 23). Breslow reported in 1970 that the 1H-NMR of cyclopropenyl cation showed a sharp singlet in the region of chemical shift δ=11.1 ppm [36]. It is far from the double bond chemical shift of cycloalkene at about 5.7 ppm. There is certainly ring current in the species. How to understand it?
Overlapping of the p-orbitals can’t explain it. Kekulé resonance structure can’t explain it either. Delocalization of electrons is also not a correct explanation, because it is difficult to imagine the state when the delocalized current passes through the positively charged C site. The C+ comes from carbon nucleus and cannot move to the center of the triangle. The flowing of electric current in ordinary conductors is caused by the repulsive force of a negative electric field on the unpaired electron orbits and the attraction of a positive electric field on the unpaired electron orbits. In the case of C3 H3 +, the positive C(1) site attracts the two parallel unpaired π-electrons in the double bond between C(2) and C(3). Once the π-orbits shift to C(3) and C(1), the C(2) becomes electron deficient. IR-spectrum shows the C3 H3+ ring mode at 1293 cm-1, equal to 3.9x1013 times (Hz) of stretching and compressing vibrations per second [37]. Then C H + appears to have equal C-C bond length of 1.373 Å. When the π-orbits constantly change positions counterclockwise, a clockwise ring current is formed. The positive charge seems to be moving, but it comes from the atomic nucleus, which cannot move. The only movable species in the system is the π-electrons. Ultimately, this is the Kekulé’s description that any two adjacent carbon atoms were connected part of time by a single bond and part of time by a double bond, as shown in Figure 23.

Figure 23: Aromatic Cyclopropenyl Cation. 1H-NMR singlet at δ=11.1 ppm. Equal Bond Length of 1.373 Å, IR Ring Mode 1293 cm-. The Flowing of Ring Current Is Caused by The Attraction Between C+ and π-Electrons.

Figure 24: Aromatic Stability of Cyclobutadiene Dication. Analogue to Cyclopropenyl Cation, the C+ Attracts the π-Electrons to Form Ring Current. C(4)+ Attracts π-Electrons Leading To Electron Deficient at C(2). When π-Electrons Moves to C(1) and C(4), Leaving C(3) Electron Deficient. Finally, Electric Current Is Generated in The Planar Ring.
Figure 24 schematically shows the cyclobutadiene dication. The removal of two π-electrons from the cyclobutadiene molecule results in the formation of the cyclobutadiene dication, C4 H4 2+, which was not experimentally characterized, due to changes positions at a speed of 1013 times per second, a ring current is formed. It looks like that the bond lengths are equalized. As long as a ring current is formed, a whirl-shaped magnetic field 4 4 considerable charge-charge repulsion arising from two units of charge over only four carbon centers [38]. On the other hand, the tetramethylcyclobutenium dication, an aromatic 2 π-electron system, has been successfully prepared by Olah’s group and was characterized by NMR [39, 40]. When we understand the mechanism of the ring current generation in cyclopropenyl cation, it is easy to understand the aromatic stability of cyclobutadiene dication.
The positively charged C(4) site attracts the two parallel unpaired π-electrons in the double bond between C(2) and C(3) during thermal vibration. Once the π-orbit shifts to C(4) and C(3), The C(2) becomes electron deficient. When the unique double bond changes positions at a speed of 1013 times per second, a ring current is formed. It looks like that the bond lengths are equalized.
As long as a ring current is formed, a whirl-shaped magnetic fieldis induced to surround the ring skeleton, resulting in aromatic stability. The planar geometry of the molecules facilitates the flowing of two parallel unpaired electron orbits (π-electrons and lone pairs of electrons) above and below the ring skeleton.
Antiaromaticity
If a molecule is cyclic, planar, and containing 4n+2 active electrons, or an odd number of double bonds and lone pairs of electrons, it has aromatic stability. If a molecule is non-planar, it does not have aromatic stability. The most challenging molecules are cyclic and planar, but contains 4n active electrons, that is, an even number of double bonds (lone pairs). These molecules are antiaromatic.

Figure 25: Aromatic, Non-Aromatic and Antiaromatic Cyclo-Seven-Membered Carbon Ring. Tropylium Cation, Planar, 4n+2 Active Electrons, Aromatic, δ=9.33 ppm, Analogue to Cyclopropenyl Cation and Cyclodutadiene Dication. Neutral Keptatriene, Nonplanar. Tropylium Anion, Planar, 4n Active Electrons, Antiaromatic.
Antiaromatic compounds are highly unstable and highly reactive. To avoid the instability of antiaromaticity, molecules may change shape, becoming non-planar.
Figure 25 is the best example of how the loss or gain of electrons in the same molecule can lead to dramatic changes in properties, namely the aromaticity, non-aromaticity, and antiaromaticity of a carbon seven-membered ring with three double bonds.
The tropylium cation has a sharp singlet 1H-NMR signal at 9.33 ppm, indicating that all seven protons are equivalent [41]. The C-C distances are equal (1.47 Å). However, the C7 H 7+ is cyclic, planar, has 4n+2 active electrons, but lacks alternating single and double bonds. How to understand its aromaticity? As with C3 H3 +, the front positively charged carbon (C+) attracts the following negatively charged π-electrons, causing the double bond to shift, leaving the electron-deficient C+ behind. By analogy, a ring current is formed along the perimeter.
The neutral cycloheptatriene has one of the carbon atoms being sp3 hybridized. The molecule is not planar, and does not satisfy the Hückel’s rule. It is non-aromatic.
The tropylium anion (C7 H7-) is cyclic, planar, has alternating single and double bonds (include lone pair), but contains even number double bonds (4n active electrons). Although it has the same configuration as tropylium cation, the tropylium anion is an unstable antiaromatic compound. The antiaromaticity has nothing to do with cation, anion or neutral molecule.
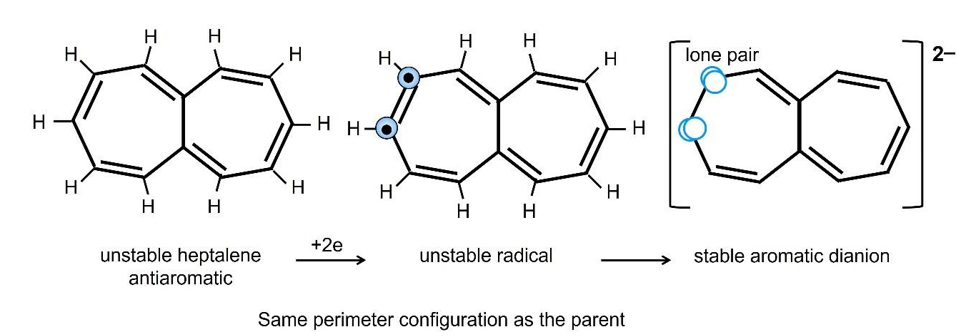
Figure 26: Neutral Antiaromatic Heptalene Changes into Aromatic Dianion, By Adding Two Extra Electrons Forming Two Lone Pairs After One of The Double Bonds Breaking, Analogue to Cyclobutadiene Dianion.
Figure 26 shows how the very unstable neutral molecule of [4n] Hückel antiaromaticity can become a very stable aromatic dianion [42]. Heptalene contains 12 active electrons or an even number of double bonds. After accepting two electrons, the compound is an extremely reactive two-electron diradical. If a double bond is opened, the two π-electrons recombine with the two new electrons to form two lone pairs of electrons, and the resulting heptalene dianion contains an odd number of double bonds and lone pairs, becoming a stable aromatic dianion. Therefore, the newly incoming electrons cannot be in a random position. They must be on either sides of a double bond to form two lone pairs. Otherwise, the aromaticity of the dianion cannot be explained.

Figure 27: Phenakistoscopic Images of The Aromatic Heptalene Dianion.
Figure 27 illustrates the resonance structures of the heptalene dianion through thermal vibration. The two unpaired parallel electrons in π-orbits and in lone pairs constantly change positions, creating a ring current along the perimeter. The ring current induces magnetic field around the skeleton to protect the molecule and stabilize it. In fact, Figure 27 shows the phenakistoscopic images mentioned by Kekulé in 1872.
Why an odd or even number of double bonds (including lone pairs) in cyclic planar molecules is the essence of aromaticity or antiaromaticity? Two unpaired electrons with parallel spins rotating in π orbits, or lone pairs, can be thought of as tiny coils of wire. With an even number of double bonds, the magnetic moments of the tiny coils alternate in an antiparallel fashion to lower the energy of the system. At the same time, the magnetic field lines of all the tiny coils form a closed loop. Such an arrangement should be stable for small magnets and normal coils. “If the magnetic moments of two bar magnets arranged in antiparallel mode, the field lines between both magnets run from one pole of one magnet to the according antipole of the other magnet.
The field line density in the space between the magnets is low, while that near the poles is high. There is an attracting force acting on both magnets (Fig. 28). [Info@HomoFaciens.de].” However, the alternating arranged electron coils are very unstable. The spins and direction of the motion of the electrons in the orbits remain unchanged. Therefore, the adjacent π-electrons will collide during the stretching and compressing vibration of the atoms, making the system unstable. This is the origin of so-called antiaromaticity, as shown in Figure 29.

Figure 28: Magnetic Field Lines of Magnets Arranged in Antiparallel Mode. The Field Line Density in The Space Between the Magnets Is Low, While That Near the Poles Is High. There is an Attracting Force Acting on Both Magnets.
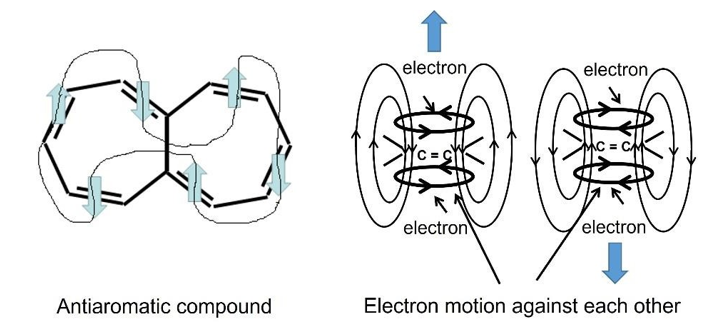
Figure 29: (a). A Closed Loop of Magnetic Moments Of the π-Orbits (coils) With Even Number. (b). The Electrons in Adjacent Coils Collide With Each Other, Making the Molecule Unstable. Under an Odd Number of Double Bonds, The Parallel Tiny Coils Always Account for Majority, Which Forces the Left and Right Adjacent Coils to Accept the Same Orientation.
With an odd number of double bonds, tiny parallel coils always dominate, forcing adjacent coils to accept the same orientation. Magnetically speaking, this arrangement is metastable. But when a ring current and ring magnetic field are formed, the molecule is stable because the electron spins all point in the same direction and the electrons move in their respective orbits in a consistent manner.
NMR data for antiaromatic compounds are not readily available. Different temperatures have a great impact on the NMR results. Here we only take [12] annulene as an example to further deepen our understanding of aromatic and antiaromatic properties.
Figure 30 shows the antiaromatic [12] annulene and its aromatic dianion. The 1H-NMR spectrum of [12] annulene has two chemical shifts at 5.9 ppm and 7.8 ppm at -170°C [43, 44]. The gas phase of [12] annulene has 1H-NMR signals at 5.856 ppm and 8.637 ppm [45]. The outer 9 protons experience typical chemical shift of cycloalkene double bond (comparing cyclohexene ring in Figure 6). The inner 3 protons experience also the strengthened magnetic field (not negative chemical shift). This shows that [12] annulene does not form a ring current.
The superimposed magnetic field inside the ring is denser than the outside, therefore, the inner protons have larger chemical shift. The natural orientation of these small coils is that their magnetic moments alternate up and down, forming a closed loop of magnetic field lines. Strong external magnetic fields force them to abandon their natural orientation and to align in one direction. But the double bonds are not completely perpendicular to the ring plane, so there will be no ring current. [12] Annulene was found to be a very unstable compound. The reason is due to the collision of adjacent orbiting electrons in the π-orbits.

Figure 30: (a). At -170°C, the Unstable Antiaromatic [12] Annulene Can Be Obtained 1H-NMR Chemical Shift at 7.8 ppm (3H) and 5.9 ppm (9H). Typical π-Orbits Induced Magnetic Field Leads to The Deshield On the Protons. (b). Aromatic Dianion of [12] Annulene Shows Typical Ring Current Induced Chemical shifts at -4.6 ppm (3H) and 6.2-7.0 ppm (9H).
The dianion of [12] annulene has the same perimeter configuration as the parent [12] annulene. It contains 14 active electrons. The 1H-NMR shows 9H 6.2 to 7.0 ppm and 3H -4.6 ppm [45, 46], typical aromatic signals like [18] annulene (12H 9.25 ppm and 6H -2.9 ppm) [31].
It can be said that in case of 4n active electrons, there are localized π-electrons and in case of 4n+2, there are flowing π-electrons. This reveals the nature of Hückel’s empirical rule.
Understanding of Aromatic Carbo-Benzene (C18H6) and Cyclo[18]Carbon (C18)
Figure 31 shows the structure of carbo-benzene (C18H6). If the 6 hydrogens are fully replaced by phenyl groups, the resulting molecule is stable. Its 1H-NMR spectrum shows that the phenyl ortho protons have larger chemical shift of 9.45 ppm compared to 1H-NMR of benzene at 7.26 ppm. This is strong evidence that the ortho proton of the phenyl group experiences the superimposed paramagnetic fields from benzene ring and the carbo-benzene ring. The other experimental facts support the aromaticity of C18Ph6 as following: average bond length of the ring d=1.332 Å. UV–vis spectroscopy λ max = 476 nm (6.3•108 Hz, ring mode) [47]. The aromatic character of the C18H6 molecule, as well as the generation of its ring currents, have long been a mystery.

Figure 31: Aromatic Carbo-Benzene (C18H6). Strong Evidence of The Existence of Large Ring Current, δ=9.49 ppm, Equal Bond Length 1.332 Å, Vibration Ring Mode 6.3•108 Hz.

Figure 32: Image in Challenges in Aromaticity: 150 Years after Kekulé’s Benzene (2015). It Remains an Unsolved Mystery to This Day (2025).
Figure 32 appeared in the themed collection: Challenges in Aromaticity: 150 Years after Kekulé’s Benzene in 2015 [48]. The C18H6 ring consists of single bonds, cumulene three double bonds and triple bonds. The covalent σ-bonds construct the ring skeleton. The cumulene three double bonds should have the electronic structure as shown in Figure 33. Mutually perpendicular π orbits separated by their respective σ-bonds are the most reasonable distribution of unpaired electrons in space and electric field. The triple bond should have the electronic structure as shown in Figure 34. Two parallel π-orbits are perpendicular to the other two parallel π-orbits. A covalent σ-bond separates them.

Figure 33: Electronic Structure of Cumulene Three Double Bonds. Two Double Bonds Are Parallel to The Ring Plane and One Double Bond Is Perpendicular to The Ring Plane (Stationary).
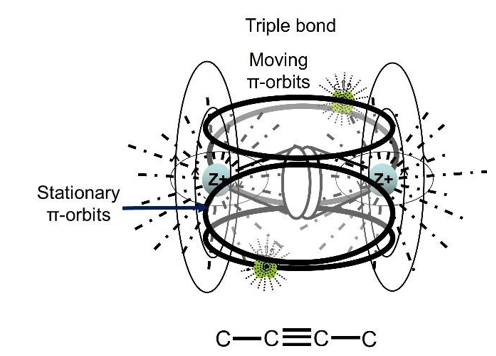
Figure 34: Electronic Structure of Triple Bond. Two π-orbits of Unpaired Electrons Are Parallel to Ring Plane and Other Two π-orbits are Perpendicular to The Ring Plane (Stationary).

Figure 35: Kekulé Oscillation Structure of Carbo-Benzene (C18H6). The Movable π-Orbits Parallel to Ring Plane Shift One Position, The Triple Bond Changes to Cumulene Three Double Bonds And Vice Versa.
If the electronic structures of cumulene and triple bonds are added to the structure in figure 32, there is an odd number of double bonds parallel to the ring plane, satisfying the Hückel's 4n+2 rule. During the thermal vibration process, the σ-bonds are constantly compressed and stretched at 6.38•108 times per second, causing the π-orbits parallel to the ring plane to shift, generating ring current similar to that of the benzene molecule. The π-orbits perpendicular to the ring plane in triple bonds and in cumulene are stationary. The C18H6 resonance structure is shown in Figure 35. The middle part of the original cumulene becomes a triple bond, and the triple bond becomes a cumulene three double bonds. This is where the aromatic stability of carbo-benzene comes from.
The understanding of the C18H6 aromatic compound leads directly to the understanding of the aromatic cyclo[18]carbon (C18).
In 1989, F. Dieterich et al. published the pioneering work in Science that they created cyclo [18] carbon from a stable organic precursor. This molecule was made up of only sp-hybridized carbon atoms. The all-sp-hybridized ring is too strain with high reactivity. It could not be structurally characterized. It is inferred based on the four valence of carbon atoms and the neutral characteristics of C18 molecule, that the ring should possess two perpendicular π-conjugated electron systems. Both systems obey Hückel’s 4n+2 rule and two orthogonal ring currents would be formed, causing double aromatic stabilization (s. Fig. 36). However, it lacks NMR experimental results to prove the existence of ring currents and lacks X-ray diffraction results to determine the equalized C-C bond length for homoatomic aromatic rings.

Figure 36: Structure of Cyclone [18] Carbon (C18) Published in Science (2019) [50]. The P-Orbitals Are Very Difficult to Explain the Possible Polyynic Or Cumulenic Structure.
In 2019, K. Kaiser et al. reported in Science that they successfully isolated C18 and visualized the molecule with AFM at 5 Kelvin on NaCl surface [50, 51]. This will greatly advance the understanding of this homoatomic ring. Whether cyclocarbons are polyynic, with alternating single and triple bonds of different lengths (D9h symmetry), or cumulenic with consecutive double bonds (D18h symmetry, s. Fig. 36) is fundamental and controversial. The AFM image showed a 9-fold symmetry, indicating a polyynic structure with defined positions of alternating triple and single bonds. Therefore, they ruled out the possibility of the presence of the cumulenic structure. However, in 2023, L. Sun et al, visualized the cumulene structure of cyclo[10]carbon and cyclo[14]carbon with the same AFM method at 4.7 Kelvin [52]. H. L. Anderson et al. mentioned in their article “A Short History of Cyclocarbons” that gas-phase electronic ultraviolet-visible spectra showed the peaks of cyclo[18]carbon and cyclo[14]carbon at 17000 cm-1 (5.1•108 Hz) and 19000 cm-1 (6.0•108 Hz) respectively [53]. This is in the same order of magnitude as the vibration frequency of aromatic carbon-benzene.
The constant oscillation of the ring provides the possibility to shift the position of the double bonds. If we introduce the electronic structures of the triple bond and the cumulene bonds in Figures 32 and 33 into C18, we can immediately see that the polyynic structure and the cumulenic structure are simply the resonance isomers during the Kakulé’s thermal vibration. Kaiser’s group reported two different AFM images of C18 (embedded in polyyne ring and cumulene ring). In fact, they had obtained the images of the two structures, but they did not pay enough attention when publishing their article. Afterwards, they further filtered the cumulene image and obtained a clear nonagon (not D18h symmetry, color AFM image), as shown in Figure 37.

Figure 37: Kaiser et al. Observed AFM Images of C18 at 5 Kelvin on NaCl Surface, Showing Both Polyynic and Cumulenic (nonagon) Structures [50]. The Electronic Structures of Triple Bond and Cumulene Bonds Correspond Well with The Observed Images.
This adds new difficulties to the understanding of C18. The nine bright bulges in first image are the triple bonds, which show clearly the polyynic structure. The second nonagon image should represent the nine π-orbits parallel to the ring plane on the outermost surface of the ring perimeter. The π-orbits perpendicular to ring plane are stationary, the same as in carbo-benzene. The moveable π-orbits satisfied four conditions of Hückel’s rule: cyclic, planar, alternating double and single bonds, and 4n+2 active electrons (or odd number double bonds). There will be no double ring-currents, because the π-orbits perpendicular to the ring plane cannot form a plane current, which do not satisfy the Hückel’s rule. The Kekulé resonance isomers is due to the thermal vibration.
The acquired time for AFM images is much longer than the thermal vibration frequency of the ring mode. The polyynic and the cumulenic rings must be frozen on the NaCl surface at 5 Kelvin. Kaiser et al. pointed out that interaction with the surface of the substate may affect the molecular structure, and the structure of C18 in vacuum might be different from that on NaCl. Up to now, C10, C12, C13, C14, C16, C18 and C20 have been visualized with AFM. If the last-step precursor contains more double bond, for example C10 and C14, the cumulenic structure was easy be detected [52]. Otherwise, the polyynic structures were captured by multiple-triple-bonds-containing precursors, such as C12, C13, C16, C18, C20 [51, 54, 55, 56].
Cyclo[N]carbons consists of N carbon atoms. N=4k+2 refers to aromatic, N=4k refers to antiaromatic. This means that N is always an even number. This classification is fine for even-numbered rings, but problematic for odd-numbered rings like C13. In 2024, F. Albrecht et al. published two AFM images of C13 in Science (Fig. 38). One AFM image shows single and triple bonds and the other image shows a polyagon [56]. They marked a lone pair of electrons in the C13 ring. Lone pairs can be thought of as small π-bonds with orbits above and below the ring and parallel to the plane of the ring. In this way, there are seven double bonds in C13 that can be shifted. which obey the Hückel’ rule.

Figure 38: Albrecht et al. Observed AFM Images of C13 at 5 Kelvin on NaCl Surface, Showing Resonance Structures [56]. The Lone Pair of Electrons Participates in Ring Current.
The above two AFM images should be the aromatic resonance structures of the Cyclo[13]carbon. When the movable π-orbits shift one position, it is the cumulenic structure, shift two steps, it is the polyynic structure. This example further illustrates that the π-bonds perpendicular to the ring plane in the triple bonds are stationary and do not participate in the ring current.
Scientists try to understand the cyclo[N]carbon by using two orthogonal arranged p-orbitals as shown in Figure 36. Since the introduction of orbital theory, it has blurred the vision of scientists.
According to report, Bohr once said to Pauling that to study the structure of the benzene ring, it is necessary to understand the structure of the double bond. That's the key point. The 1H-NMR revealed the double bond structure clearly (s. Fig. 2). Two parallel (spins in the same direction) unpaired electrons are in the double bond. The movement of the two electrons describes two π-orbits with magnetic property. The shift of the π-orbits generates ring current. The ring current induces extra magnetic field around the cyclic skeleton, to stabilize the aromatic molecules.
The orbital theory loses the electron spin. The delocalization theory loses the π-orbits. The electron cloud hypothesis does not take into account that electrons have charge, mass, and electron spin. The Schrödinger theory means that the orbitals are the possibility to find any electron in an atom in any specific region around the nucleus. His theory ignores a simple fact, that once the electron appears in the position of the nucleus, the electron will be sucked into the nucleus.
Conclusion
In this work, the aromaticity of various cyclic compounds has been comprehensively analyzed. Kekulé’s oscillation idea is the soul of the aromaticity. The covalent σ-bonds constructs the ring skeleton. The π-electrons in the double bonds can shift along the perimeter of the aromatic ring due to thermal vibration to create ring current and additional ring magnetic field. Lone pairs of electrons can be treated as small π-electrons. There are stationary π-orbits in triple bonds and in cumulenic bonds in cyclic C18H6 and C18 molecules, which are perpendicular to the aromatic ring plane.
The Huckel’s rule, (planar, cyclic, alternating single and double bond, 4n+2 active electrons or odd number double bonds), is suitable for judging aromatic compounds. In addition, the repulsion between π-electrons or the attraction between C+ and π-electrons, both can trigger the flow of double bonds. As a result, any two adjacent carbon atoms were connected part of time by a single bond and part of time by a double bond.
Acknowledgements
The author would like to thank Dr. Thomas VON LARCHER, Senior Editor, Research Publishing, Springer Nature, for his many suggestions in revising this article.
Funding
Not Applicable.
Clinical Trial Number
Not Applicable.
Ethics, Consent to Participate, and Consent to Publish declarations: not applicable.
References
- Faraday, M. (1825). XX. On new compounds of carbon and hydrogen, and on certain other products obtained during the decomposition of oil by heat. Philosophical Transactions of the Royal Society of London, (115), 440-466.
- Kaiser, R. (1968). “Bicarburet of hydrogen”. Reappraisal of the discovery of benzene in 1825 with the analytical methods of 1968. Angewandte Chemie International Edition in English, 7(5), 345-350.
- Mitscherlich, E. (1834). Über das Benzol und die Säuren der Oel-und Talgarten. Ann. Pharm, 9, 39-48.
- Martín, N., & Scott, L. T. (2015). Challenges in aromaticity: 150 years after Kekulé's benzene. Chemical Society Reviews, 44(18), 6397-6400.
- Kekulé, F. A. (1865). Sur la constitution des substances aromatiques. Bulletin mensuel de la Société Chimique de Paris, 3, 98.
- Kekulé, F. A. (1866). Untersuchungen über aromatischeverbindungen. Ann. Chem. Pharm. 137, 129–196.
- Kekulé, F. A. (1872). Ueber einige condensationsproducte des aldehyds. Justus Liebigs Annalen der Chemie, 162(1), 77-124.
- Lonsdale, K. (1929). The structure of the benzene ring in C6 (CH3)6. Proceedings of the Royal Society of London. Series A, Containing Papers of a Mathematical and Physical Character. 123 (792), 494-515.
- Lonsdale, K. (1928). The structure of the benzene ring.Nature, 122(3082), 810-810.
- Mark, H. & Wierl, R. (1930). Neuere Ergebnisse der Elektronenbeugung. Die Naturwissenseh. 18, 778-786 (1930).
- Wierl, R. (1931). Electronenbeugung und Molekülbau. Ann.d. Phys. 200 (5), 521-564.
- Tamagawa, K., Iijima, T., & Kimura, M. (1976). Molecular structure of benzene. Journal of Molecular Structure, 30(2), 243-253.
- Hückel, E. (1931). Quantentheoretische Beiträge zum benzol problem. I. Die elektronenkonfiguration des Benzols und verwandter Verbindungen. Zeitschrift für Physik, 70, 204-286.
- Hückel, E. (1931). Quanstentheoretische beiträge zum benzolproblem: II. quantentheorie der induzierten polaritäten. Zeitschrift für Physik, 72(5), 310-337.
- Hückel, E. (1932). Quantentheoretische Beiträge zum Problem der aromatischen und ungesättigten Verbindungen.III. Zeitschrift für Physik, 76(9), 628-648.
- Bloch, F. (1929). Über die quantenmechanik der elektronen in kristallgittern. Zeitschrift für physik, 52(7), 555-600.
- Schrödinger, E. (1926). Quantisierung als Eigenwertproblem; von Erwin Schrödinger. Annalen der Physik, 384(4), 361-377.
- Pauling, L., & Wheland, G. W. (1933). The nature of the chemical bond. V. The quantumâ?mechanical calculation of the resonance energy of benzene and naphthalene and the hydrocarbon free radicals. The Journal of Chemical Physics,1(6), 362-374.
- Cooper, D. L., Gerratt, J., & Raimondi, M. (1986). The electronic structure of the benzene molecule. Nature, 323(6090), 699-701.
- Maddox, J. (1987). Benzene's electron structure. Nature, 327(6123), 551.
- Schultz, P. A., & Messmer, R. P. (1987). Are there π bonds in benzene? Physical Review Letters, 58(23), 2416.
- Gross, L., Mohn, F., Moll, N., Liljeroth, P., & Meyer, G. (2009). The chemical structure of a molecule resolved by atomic force microscopy. Science, 325(5944), 1110-1114.
- Liu, Y., Kilby, P., Frankcombe, T. J., & Schmidt, T. W. (2020). The electronic structure of benzene from a tiling of the correlated 126-dimensional wavefunction. NatureCommunications, 11(1), 1210.
- Thomson, J. J. (1906). Carriers of negative electricity. NobelLecture, December, 11(1906), 1901-1921.
- Millikan, R. A. (1913). On the elementary electrical chargeand the Avogadro constant. Physical Review, 2(2), 109.
- Shimajiri, T., Kawaguchi, S., Suzuki, T., & Ishigaki, Y. (2024). Direct evidence for a carbon–carbon one-electron σ-bond. Nature, 634(8033), 347-351.
- Pauling, L. (1931). The nature of the chemical bond. II. The one-electron bond and the three-electron bond. Journal of the American Chemical Society, 53(9), 3225-3237.
- Streitwieser, A. & Heathcock, C. H. (1986). “Organische Chemistrie”. Weinheim: VCH.
- Goudsmit, S., & Uhlenbeck, G. E. (1926). Spinning electrons and the structure of spectra. Nature, 117(2938), 264-265.
- Lowry, T. H. & Richardson, K. S. Mechanism and theory in organic chemistry (1987). (3rd ed.). New York: Harper & Row. ISBN 0060440848. OCLC 14214254.
- Gilles, J. M., Oth, J. F. M., Sondheimer, F., & Woo, E. P. (1971). Unsaturated macrocyclic compounds. Part LXXXIII. A quantitative study of the conformational mobility of [18] annulene. Journal of the Chemical Society B: Physical Organic, 2177-2186.
- Lee, V. Y., Takanashi, K., Kato, R., Matsuno, T., Ichinohe, M., & Sekiguchi, A. (2007). Heavy analogues of the 6π-electron anionic ring systems: cyclopentadienide ion and cyclobutadiene dianion. Journal of organometallic chemistry, 692(13), 2800-2810.
- Breton, G. W. (1997). Generation and Observation of the Cyclopentadienyl Anion: A Negatively Charged Aromatic Molecule. The Chemical Educator, 2(6), 1-8.
- Bachmann, S., Gernert, B., & Stalke, D. (2016). Solution structures of alkali metal cyclopentadienides in THF estimated by ECC-DOSY NMR-spectroscopy (incl. software). Chemical Communications, 52(87), 12861-12864.
- Matsuo, T., & Sekiguchi, A. (2004). Cyclobutadiene dianion.Bulletin of the Chemical Society of Japan, 77(2), 211-226.
- Breslow, R., & Groves, J. T. (1970). Cyclopropenyl cation. Synthesis and characterization. Journal of the American Chemical Society, 92(4), 984-987.
- Marimuthu, A. N., Sundelin, D., Thorwirth, S., Redlich, B., Geppert, W. D., & Brünken, S. (2020). Laboratory gas-phasevibrational spectra of [C H ]+ isomers and isotopologues by IRPD spectroscopy. Journal of Molecular Spectroscopy, 374,111377.
- Firme, C. L., Antunes, O. A. C., & Esteves, P. M. (2007). Electronic nature of planar cyclobutenyl dication derivatives. The Journal of Physical Chemistry A, 111(46), 11904-11907.
- Olah, G. A., Bollinger, J. M., & White, A. M. (1969). Stable carbonium ions. LXXXIX. Tetramethylcyclobutenium dication, an aromatic 2 π-electron system. Journal of the American Chemical Society, 91(13), 3667-3669.
- Olah, G. A., & Staral, J. S. (1976). Novel aromatic systems. 4. Cyclobutadiene dications. Journal of the American Chemical Society, 98(20), 6290-6304.
- Zahra, F. T., Saeed, A., Mumtaz, K., & Albericio, F. (2023).Tropylium Ion, an Intriguing Moiety in Organic Chemistry.Molecules, 28(10), 4095.
- Oth, J. F. M., Müllen, K., Königshofen, H., Wassen, J., & Vogel, E. (1974). The dianion of heptalene. Helvetica Chimica Acta, 57(8), 2387-2398.
- Braten, M. N., Castro, C., Herges, R., Köhler, F., & Karney, W.L. (2008). The [12] annulene global minimum. The Journal of Organic Chemistry, 73(4), 1532-1535.
- Oth, J. F. M. (1971). Conformational mobility and fast bond shift in the annulenes. Pure and Applied Chemistry, 25(3), 573-622.
- Goyary, S., Sarmah, M. J., Goswami, H. P., & Nath, N. (2024). Solvent-induced 1H NMR chemical shifts of annulenes. Computational and Theoretical Chemistry, 1236, 114601.
- Oth, J. F. M., & Schröder, G. (1971). Annulenes. Part XII. The dianion of [12] annulene. Journal of the Chemical Society B: Physical Organic, 904-907.
- Zou, C., Lepetit, C., Coppel, Y., & Chauvin, R. (2006). Ring carbo-mers: From questionable homoaromaticity to bench aromaticity. Pure and applied chemistry, 78(4), 791-811.
- Cocq, K., Lepetit, C., Maraval, V., & Chauvin, R. (2015). “Carbo-aromaticity” and novel carbo-aromatic compounds. Chemical Society Reviews, 44(18), 6535-6559.
- Diederich, F., Rubin, Y., Knobler, C. B., Whetten, R. L., Schriver, K. E., Houk, K. N., & Li, Y. I. (1989). All-carbon molecules: evidence for the generation of cyclo [18] carbonfrom a stable organic precursor. Science, 245(4922), 1088- 1090.
- Kaiser, K., Scriven, L. M., Schulz, F., Gawel, P., Gross, L., & Anderson, H. L. (2019). An sp-hybridized molecular carbon allotrope, cyclo [18] carbon. Science, 365(6459), 1299-1301.
- Scriven, L. M., Kaiser, K., Schulz, F., Sterling, A. J., Woltering,S. L., Gawel, P., ... & Gross, L. (2020). Synthesis of cyclo [18] carbon via debromination of C18Br6. Journal of the American Chemical Society, 142(30), 12921-12924.
- Sun, L., Zheng, W., Gao, W., Kang, F., Zhao, M., & Xu, W. (2023). On-surface synthesis of aromatic cyclo [10] carbon and cyclo [14] carbon. Nature, 623(7989), 972-976.
- Anderson, H. L., Patrick, C. W., Scriven, L. M., & Woltering,S. L. (2021). A short history of cyclocarbons. Bulletin of the Chemical Society of Japan, 94(3), 798-811.
- Sun, L., Zheng, W., Kang, F., Gao, W., Wang, T., Gao, G., & Xu, W. (2024). On-surface synthesis and characterization of anti-aromatic cyclo [12] carbon and cyclo [20] carbon. Nature Communications, 15(1), 7649.
- Gao, Y., Albrecht, F., RonÄeviÄ?, I. et al. On-surface synthesis of a doubly anti-aromatic carbon allotrope. Nature 623, 977- 981 (2023).
- Albrecht, F., RonÄeviÄ?, I., Gao, Y., Paschke, F., Baiardi, A., Tavernelli, I., ... & Gross, L. (2024). The odd-number cyclo [13] carbon and its dimer, cyclo [26] carbon. Science, 384(6696), 677-682.