
International Journal of Women's Health Care(IJWHC)
ISSN: 2573-9506 | DOI: 10.33140/IJWHC
Impact Factor: 1.011

Case Report - (2021) Volume 6, Issue 2
Nerve Sparing Reduction Clitoroplasty in a Case of Congenital Adrenal Hyperplasia
2HOD, Dept of Gen surgery, Khaja Banda Nawaz Institute of Medical Sciences, Gulbarga, India
Received Date: Apr 21, 2021 / Accepted Date: Apr 22, 2021 / Published Date: May 03, 2021
Copyright: ©Bibi Zainab, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Citation: Bibi Zainab, Mohammad Moinuddin. (2021). Nerve Sparing Reduction Clitoroplasty in a Case of Congenital Adrenal Hyperplasia. Int J Women's Health Care, 6(2), 161-164.
Abstract
Adrenogenital syndrome, or Congenital Adrenal Hyperplasia, is caused by a congenital insufficiency of the enzyme 21-hydroxylase, which is responsible for converting cortisol into cholesterol. Because of virilizing effect of androgens and its over- production, girls develop clitoral hypertrophy or Clitoromegaly. Clitoromegaly is an embarrassing condition, causing psychological stress to young girls and, hence requiring intervention. The goals of clitoroplasty are to achieve normal genital anatomy and to preserve tactile sensation with a satisfactory sexual response. We present a case of Adrenogenital syndrome with Clitoromegaly managed by reduction Clitoroplasty, preserving the dorsal neurovascular bundle and extensive network of nerves around the corpora to the glans and there by preserving the tactile sensation of the clitoris.
Keywords
Adrenal Hyperplasia, Ambiguous Genitalia, Clitoris, Clitoromegaly, Nerve Sparing Clitoroplasty
Introduction
This is a familial disease transmitted by a recessive gene. If any couple has had one affected child, a subsequent baby has a 1 in 4 chance of having the same disability.
The cause of the disease is an inherent enzymatic error in the ad- renal cortex, which is unable to complete the normal biosynthesis whereby progesterone is converted through hydroxy progesterone to cortisol. There are several levels at which arrest can occur, with consequent variations in the effects. The most common enzymatic defects are those of 21-hydroxylase, 11 β-hydroxylase and 3 β-hy- droxysteroid dehydro- genase.
Figure 1: The relations between the hypothalamic-pituitary sys- tem and the adrenal cortex in congenital adrenal hyperplasia. Synthesis of corticoids by the adrenal is blocked, usually at the hydroxyprogesterone link in the chain. The excretion product of this substance, pregnanetriol, therefore appears in large amounts in the urine and liquor amnii. Absence of the corticoid inhibitory ef- fect on the hypothalamus and pituitary allows an excessive output of ACTH and consequent overstimulation of the adrenal cortex. The result is increased blood levels and secretion of androgens and sometimes other products, and a rise in the urinary excretion of 17-ketosteroid.
After birth the fundamental metabolic upset continues, so the ex- cessive adrenal cortical function causes physical and sexual pre- cocity which, in the female child, is of the virile pattern. Pubic and axillary hair appear, and the voice deepens by the age of 2-4 years. Affected children at first grow quickly but the epiphyses of their long bones close at the age of 8 or 9 years.
The ovaries, although normally formed, do not function; the uter- us remains infantile and fails to menstruate; hirsutism becomes a problem.
In the more severe forms of the disease, salt-wasting and shock may also be present at birth. Very mild, nonclassical disease may present only at puberty. When male foetuses suffer from congenital adrenal hyperplasia, their precocious masculinity is accentuated to produce the ‘infant Hercules.
The abnormality of the vulva can readily be corrected by plastic surgery. Exposure of the vagina can be deferred until puberty but, to avoid a psychological reaction, the large phallus should be re- moved before the age of 5 years. Early diagnosis and treatment are important, otherwise voice changes, persistent hirsutism and premature closure of epiphyses leave permanent stigmata. With steroid therapy, ovarian and uterine functions become normal, and fertility is possible. The disorder is transmitted to the offspring as a monogenic autosomal recessive trait. Psychological counselling is required both for the parents and the child.
Several procedures have been described to reduce the size of the phallus, including the amputation of the hypertrophied clitoris (Cl- itorectomy), which used to be practiced very often in the past [1,2]. However, the preservation of the sensitive glans (clitoroplasty) has become the most chosen technique by investigators [3,4].
A clitoral index of >35 mm2 (length × width) is called Clitoro- megaly [2,3]. The nerves and vessels form an extensive network around the end of corporeal bodies with an extensive network of nerves to the glans distally [1].
Optimal sexual function after surgical correction of clitoral hyper- trophy requires adequate innervation and vascular supply to the glans clitoris. These goals are accomplished by shortening the cor- pora cavernosa, while preserving the neurovascular supply to the glans.
Several clitoroplasty methods have been reported, but few de- scribe preservation of dorsal and ventral neurovascular bundles in sexually mature women. We present a case of clitoromegaly in a case of congenital adrenal hyperplasia treated with clitoroplasty, where the dorsal neurovascular bundle was preserved along with the extensive network of nerves around the end of corporeal bodies to the glans.
Case Report
We report a case of a 17-year-old female patient presenting with primary amenorrhea, change in voice and delayed growth, fascial hair.
The child was born out of non consangious marriage. The mother had never been treated with any drugs during pregnancy or en- countered hormonal exposure in utero and she had no signs of an- drogen excess such as hirsutism, alopecia or clitoral hypertrophy (maternal virilization).
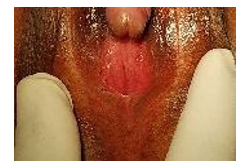
On clinical examination the patient had hirsutism on the face (up- per lips) and had a masculine look. Height was stunted for age 142cm, axillary pubic hair growth, breast bud was present but not developed was present. Genital examination revealed a phallus measuring 4cm, urethra not visualized, vaginal opening seen.
Hormone analysis shows 17-OH Progesterone 18.4ng/mL. Serum Testosterone 189ng/dL.
On Ultrasonography prepubertal sized uterus and bilateral ovaries were present with multiple follicles
Xray hand bone age correlates with the birth age, all calyceal bone ossification centers present.
Surgical Procedure
Surgery was done in the lithotomy position. A stay suture was placed on the glans for traction using 4–0 monofilament. Incision was made on the prepuce skin 2-3 mm proximal and parallel to the corona of the glans, extending 270° on the dorsolateral surface of the prepuce. The ventral connection of the skin was preserved.
The skin was retracted over the phallus toward the base to expose Buck’s fascia, but the ventral tissue was preserved. Tourniquet was applied to
the base using a vascular loop.
Two longitudinal incisions were made on both sides of the phallus, lateral to the dorsal neurovascular bundle through Buck’s fascia up to the corpora cavernosa.

The dorsal neurovascular bundle was isolated and lifted off the phallic shaft using a vascular loop. Ventral urogenital tissue was isolated and secured with a vessel loop, and vessel loop previously placed around the base of the phallic shaft as a tourniquet was tightened to minimize bleeding. The dissection was carried proxi- mally to the bifurcation of the corpora. The two crura were identi- fied, clamped and the mid body of the corpora resected.
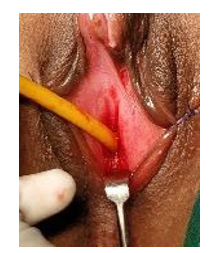
The proximal corpora were closed with 4–0 Vicryl for haemosta- sis. The base of the glans was sutured to the divided proximal cor- pora with 4–0 Vicryl interrupted sutures. After haemostasis was confirmed, the skin was closed with 4–0 vicryl interrupted sutures to re-approximate the labia minor and the clitoris
Histopathological report-normal corporal tissue.
Follow-up: The patient is under follow-up since operation. There were no early and late postoperative complications. Sensation was normal and the patient was satisfied with the appearance
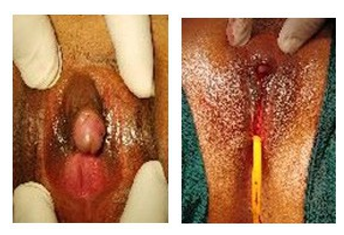
Discussion
Surgical methods for correction of clitoral hypertrophy were first described in 1934 by Young who performed an operation for cli- toral reduction in a child with congenital adrenal hyperplasia [5]. Young preserved the glans clitoris, but the glans later sloughed leaving granulation tissue [4]. In 1954, Jones and Jones report- ed a modification to Young’s operation, but the remaining erectile tissue was painful, leading to the development of procedures for clitoral reduction (removal of a segment of the corpora bodies) that included preservation of clitoral innervation [1,6].
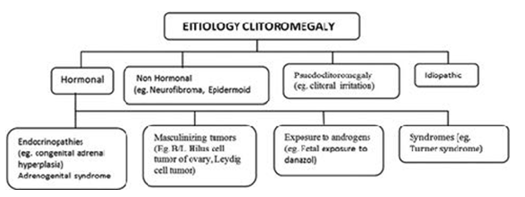
Treatment includes medical therapy to treat the underlying cause and surgical clitoroplasty safeguarding the neurovascular pedicles for preservation of sexual arousal function [5].
Knowledge of anatomy is crucial. There is a dorsal neurovascular pedicle from the pudendal nerve along with an extensive network of nerves to the glans [7]. The contribution to different parts of the clitoris to sexual sensitivity, arousal and orgasm remains unclear [8]. Major difference between male and female external genita- lia is urethra. Cross-sectional anatomy of the clitoris is same as the penis but has no urethra and corpora sponsiosum [Figure 10]. Surgical methods for correction of clitoral hypertrophy were first described in 1934 by Young.
In the female, the normal glans is small with thin corpora cav- ernosa. The goal of a reducing clitoroplasty is to create a female phenotype to satisfy the parents and permit the development of a good gender identity in the patients.
In 1961, Lattimer described the first clitoroplasty by relocation and recession of the hypertrophied clitoris through a subcutaneous tun- nel as an alternative to clitoral [9]. amputation (clitorectomy) [2]. Afterward, Kumar et al. proposed a partial excision of the corpora cavernosa after dissection of the dorsal neurovascular bundle [10]. Since then, reduction clitoro- plasty by excision has been the most accepted and widely used technique.
The innervation and vascularization of the glans are ensured by 2 dorsal neurovascular bundles that pass at the 11- and 1 o’clock positions. Distally, the nerves and vessels penetrate the glans and form an extensive network around the end of corporeal bodies and sometimes the urethra. I believe that the presence of the nerves surrounding the tunica with many perforating branches entering the dorsal aspect of the corporeal bodies and glans does not forbid the dissection of the dorsal neurovascular bundles, because we ex- cise a large area of the corporeal bodies, and the study by Gearhart et al. demonstrated preservations of a genital electromyographic response at the glans after stimulating the dorsal neuro- vascular bundle, both before and after clitoral reduction [11,12].
With this operation, the complete mobilization of the glans with its neurovascular bundles facilitates the surgery and makes it pos- sible to trace the part that needs to be excised. This gives results in a symmetric reduction. The reduction of the size of an enlarged glans is essential, and the splitting of the glans allows us to effect a harmonious reduction of the glans by excision of the distal edges [13,14].
References
- Acimi S (2008) Clitoroplasty: A variant of the technique. Urology 72: 669-671.
- Young HH (1937) Genital abnormalities. In: Hermaphrodit- ism and Re- lated Adrenal Diseases. Baltimore: Williams & Wilkins 119.
- Gross RE, Randolph J, Crigler JF Jr (1966) Clitorectomy for sexual abnormalities: Indication and technique. Surgery 59: 300-308.
- Young HH (1937) Genital abnormalities, hermaphroditism and related adrenal disease. Baltimore, MD: Williams & Wilkins 103-105.
- Tuteja N, Saluja S, Jain SK, Yadav A, Agarwal LD, et al. (2014) Congenital Idiopathic Isolated Clitoromegaly. Int J App Basic Med Res 4: 192-194.
- Rajfer J, Ehrlich RM, Goodwin WE (1982) Reduction clitoro- plasty via ventral approach. J Urol 128: 341-343.
- O’Connell HE, Sanjeevan KV, Hutson JM (2005) Anatomy of the clitoris. J Urol 174: 1189-1195
- Minto CL, Liao LM, Woodhouse CR, Ransley PG, Creighton SM (2003) The effect of clitoral surgery on sexual outcome in individuals who have intersex conditions with ambiguous genitalia: A cross-s. ectional study. Lancet 361: 1252-1257.
- Lattimer JK (1961) Relocation and recession of the enlarged clitoris with preservation of the glans: An alternative to ampu- tation. J Urol 86: 113-116.
- Kumar H, Kiefer H, Rosenthal IE, S S Clark (1974) Clitoro- plasty: Experience during a 19-year period. J Uro. 111: 81-84.
- Baskin LS, Erol A, Li YW, W H Liu, E Kurzrock, et al. (1999) Anatomical studies of the human clitoris. J Urol 162: 1015- 1020.
- Gearhart JP, Burnett A, Owen JH (1995) Measurement of pudendal evoked potentials during feminizing genitoplasty: Technique and application. J Urol 153:486-487.
- Huma Z, Crawford C, New MI (1996) Congenital Adrenal Hyper- plasia. Clin Pediatr Endocrin, ed Ch GD Brook.
- Nigam A, Prakash A, Sarema P, Yadav R, Raghunandan C (2011) Hirsutism and abnormal genitalia. JIACM 12: 46-48.