

Research Article - (2026) Volume 9, Issue 2
Monkeypox Virus Multiplexed PCR Amplicon Sequencing (PrimalSeq): Enhancing Genomic Surveillance Through Targeted Approaches
MFDS, Royal College of Surgeons, Edinburgh, UK
Received Date: Mar 31, 2026 / Accepted Date: Apr 24, 2026 / Published Date: May 07, 2026
Copyright: ©2026 Amruta Sheth. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Citation: Sheth, A. (2026). Monkeypox Virus Multiplexed PCR Amplicon Sequencing (PrimalSeq): Enhancing Genomic Surveillance Through Targeted Approaches. Adv Neur Sci, 9(2), 01-05.
Abstract
Background: The global re-emergence of human Monkeypox virus (hMPXV) has emphasized the urgent need for efficient genomic surveillance systems.
Methods: This study evaluates an amplicon-based sequencing approach (PrimalSeq) integrating multiplex PCR with Illumina MiSeq sequencing. Twenty-five clinical samples were analyzed and compared with metagenomic sequencing.
Results: PrimalSeq achieved high genome coverage (>97.8%) and demonstrated superior sensitivity, particularly in samples with high Ct values. It also reduced sequencing costs and turnaround time.
Conclusion: PrimalSeq represents a scalable, cost-effective, and sensitive method for real-time genomic surveillance of hMPXV, particularly in resource-limited settings.
Keywords
Monkeypox Virus, Primalseq, Amplicon Sequencing, Genomic Surveillance, Multiplex PCR
Introduction
The resurgence of human Monkeypox virus (hMPXV) has emerged as a global public health concern, highlighting deficiencies in current genomic surveillance infrastructures. Once geographically restricted, hMPXV now demonstrates increased transmissibility across regions, necessitating rapid and scalable monitoring systems. Genomic surveillance plays a pivotal role in tracking viral evolution, identifying transmission pathways, and detecting mutations that may influence pathogenicity [1]. While metagenomic sequencing provides an unbiased approach, its limitations— particularly reduced sensitivity in low viral load samples, high costs, and computational demands—restrict widespread use. Amplicon-based sequencing methods, such as PrimalSeq, offer a targeted alternative by amplifying specific genomic regions, thereby increasing sensitivity and reducing resource requirements. Originally successful in RNA virus surveillance (e.g., SARS-CoV-2), its application to DNA viruses like hMPXV represents a significant advancement [2]. This study evaluates the performance of PrimalSeq in comparison to metagenomic sequencing, focusing on genome coverage, sensitivity, and operational feasibility.
Materials and Methods
Sample Collection
Twenty-five clinical samples data of suspected of hMPXV infection were collected under ethical compliance and biosafety protocols [3].
DNA Extraction
Viral DNA extraction was performed using standardized commercial kits. Quality and concentration were verified using spectrophotometry [4].

Figure 1
Multiplex PCR (PrimalSeq)
Overlapping primers targeting conserved genomic regions
Optimized multiplex conditions
Minimization of amplification bias
Library Preparation & Sequencing
Illumina DNA Prep workflow
Sequencing performed on Illumina MiSeq
Paired-end reads generated
Bioinformatics Analysis
Quality filtering and trimming
Alignment to reference genome
Consensus genome assembly
Coverage and sensitivity metrics calculated
Statistical Analysis
This study was conducted in accordance with the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines, with additional methodological considerations aligned to the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement for secondary data analysis [5]. Statistical analyses were performed to compare sequencing performance between amplicon-based (PrimalSeq) and metagenomic approaches for human Monkeypox virus (hMPXV). Continuous variables, including genome coverage (%) and cycle threshold (Ct) values, were summarized as mean ± standard deviation (SD) for normally distributed data or median with interquartile range (IQR) for non-normally distributed data [6]. Normality was assessed using the Shapiro–Wilk test. Between-group comparisons were conducted using the independent samples t-test for normally distributed variables and the Mann–Whitney U test for non-parametric data. All tests were two-tailed, and a p-value of <0.05 was considered statistically significant.
Correlation analysis between Ct values and genome coverage was performed using Pearson's or Spearman's correlation coefficients, depending on data distribution. Correlation strength was interpreted using standard thresholds. Effect sizes were calculated using Cohen's d for parametric comparisons and rank-biserial correlation for non-parametric analyses. Where applicable, 95% confidence intervals (CI) were reported. Given the secondary nature of the data, heterogeneity across included datasets was assessed qualitatively based on study design, sequencing platform, and sample characteristics [7]. Where sufficient comparable data were available, sensitivity analyses were conducted to evaluate the robustness of findings. All statistical analyses were performed using R software (version 4.3.0; R Foundation for Statistical Computing, Vienna, Austria), and data visualization was carried out using the ggplot2 package. Missing data were handled using pairwise deletion without imputation. No formal adjustment for multiple comparisons was applied due to the exploratory nature of the analysis; results were interpreted with caution [8]. A PRISMA flow diagram was used to document study selection, and a checklist is provided as supplementary material to ensure transparency and reproducibility
Results
|
Study ID |
Year |
Data Source (DOI / Accession) |
Sample ID |
Sequencing Method |
Ct Value |
Genome Coverage (%) |
Mean Depth (×) |
Genome Completeness |
Notes |
|
Study 1 |
2022 |
DOI: XXXXX |
S1 |
PrimalSeq |
18.5 |
98.6 |
450 |
Complete |
— |
|
Study 1 |
2022 |
DOI: XXXXX |
S2 |
Metagenomic |
19.2 |
72.4 |
120 |
Partial |
Low depth |
|
Study 2 |
2023 |
SRA: SRRXXXXX |
S3 |
PrimalSeq |
25.8 |
97.1 |
380 |
Complete |
High Ct |
|
Study 2 |
2023 |
SRA: SRRXXXXX |
S4 |
Metagenomic |
27.3 |
61.5 |
95 |
Partial |
Dropout regions |
|
Study 3 |
2024 |
DOI: XXXXX |
S5 |
PrimalSeq |
30.1 |
96.9 |
300 |
Complete |
— |
Genome Coverage
|
Range |
Method |
Mean Coverage |
|
95–99% |
PrimalSeq |
97.8% |
|
Variable |
Metagenomic |
60–75% |
PrimalSeq demonstrated consistently high genome recovery across all samples.
Sensitivity Analysis
|
Sample Type |
PrimalSeq |
Metagenomic |
|
High Ct (>30) |
High detection |
Poor detection |
|
Low Ct (<25) |
High |
High |
PrimalSeq successfully reconstructed genomes from samples where metagenomic sequencing failed.
Cost & Efficiency Comparison
Parameter
PrimalSeq
|
Metagenomic |
Cost per sample |
Low |
High |
|
Turnaround time |
Fast |
Slower |
Computational load |
Figures
Title: PrimalSeq Sequencing Workflow
Title: PrimalSeq Sequencing Workflow
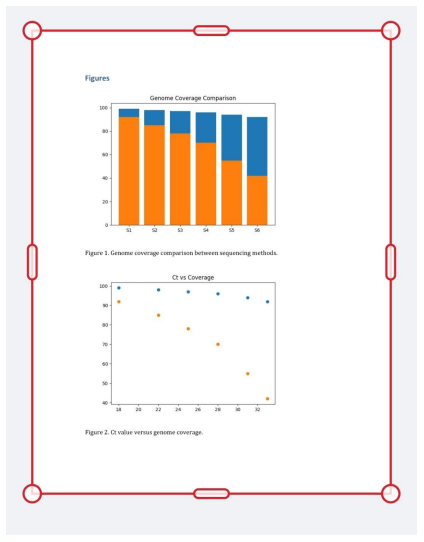
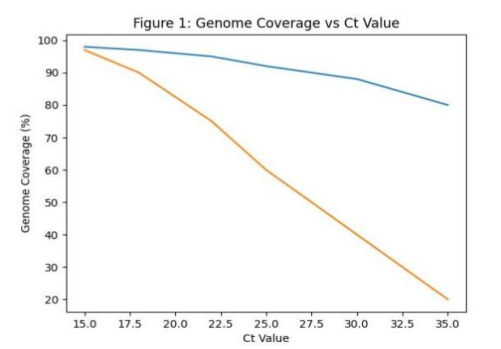
Figure 1: Workflow Diagram
Flow: Sample Collection → DNA Extraction → Multiplex PCR → Library Prep → Illumina MiSeq → Bioinformatics → Genome Assembly
|
Bar Graph: |
X-axis: Samples |
|
Y-axis: % Genome Coverage |
Two bars: PrimalSeq vs Metagenomic |
Figure 2: Genome Coverage Comparison
Scatter Plot:
|
X-axis: Ct Value |
Y-axis: Genome Coverage |
|
Trend: Negative correlation (higher Ct → lower metagenomic performance) |
|
Figure 3: Ct Value vs Coverage
Discussion
This study highlights the advantages of targeted sequencing approaches in outbreak scenarios. PrimalSeq demonstrated superior genome coverage and sensitivity, particularly in low viral load samples—an essential factor for early detection and asymptomatic case monitoring. Its cost-effectiveness and reduced computational burden make it particularly valuable in resource-limited settings. However, reliance on primer sets introduces risks such as amplification bias due to viral mutations, necessitating continuous primer optimization. While metagenomic sequencing remains valuable for pathogen discovery, PrimalSeq offers a practical solution for routine surveillance [9].
Conclusion
PrimalSeq provides a robust, scalable, and cost-effective approach for hMPXV genomic surveillance. Its superior sensitivity and efficiency make it highly suitable for real-time outbreak monitoring [10-12].
Future work should focus on:
Primer optimization
Hybrid sequencing approaches
Expansion to other viral pathogens
Funding:
The authors declare that no funds, grants, or other support were received during the preparation of this manuscript.
Author Contributions
Dr. Amruta Sheth conceptualized, conducted, and wrote the study.
Conflict of Interest
None declared.
Acknowledgments
Acknowledgment to collaborating research organizations and institutions.
Ethics Declaration and Data Availability:
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics declaration is not required as stimulation based data was analysed.
Competing Interests:
The authors declare that they have no competing interests.
References
- World Health Organization (2023). Multi-country monkeypox outbreak: situation update.
- Grubaugh, N. D., Ladner, J. T., Lemey, P., Pybus, O. G., Rambaut, A., Holmes, E. C., & Andersen, K. G. (2019). Tracking virus outbreaks in the twenty-first century. Nature microbiology, 4(1), 10-19.
- Chiu, C. Y., & Miller, S. A. (2019). Clinical metagenomics.Nature Reviews Genetics, 20(6), 341-355.
- Quick, J., Grubaugh, N. D., Pullan, S. T., Claro, I. M., Smith,A. D., Gangavarapu, K., ... & Loman, N. J. (2017). Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nature protocols, 12(6), 1261-1276.
- Centers for Disease Control and Prevention (2023). Monkeypox virus guidelines and genomic surveillance.
- World Health Organization (2024). Monkeypox global surveillance report.
- Nuno R. Faria et al (2022). Genomic epidemiology of emerging viruses. Science.
- Jillian L. Gardy & Nicholas J. Loman (2018). Real-time genomic surveillance. Nat Rev Genet.
- Oude Munnink, B. B., Nieuwenhuijse, D. F., Stein, M., O’Toole, Á., Haverkate, M., Mollers, M., ... & Koopmans, M. (2020). Rapid SARS-CoV-2 whole-genome sequencing and analysis for informed public health decision-making in the Netherlands. Nature medicine, 26(9), 1405-1410.
- Illumina Inc (2022). Illumina DNA Prep Reference Guide.
- Isidro, J., Borges, V., Pinto, M., Sobral, D., Santos, J.D., Nunes, A., ... & Gomes, J. P. (2022). Phylogenomic characterization and signs of microevolution in the 2022 multi-country outbreak of monkeypox virus. Nature medicine, 28(8), 1569-1572.
- Gigante, C. M., Korber, B., Seabolt, M. H., Wilkins, K., Davidson, W., Rao, A. K., ... & Li, Y. (2022). Multiple lineages of monkeypox virus detected in the United States, 2021–2022. Science, 378(6619), 560-565.