
Journal of Pathology and Laboratory Medicine(JPLM)



Editorial Article - (2023) Volume 1, Issue 1
Maraud and Gizzard-Hepatosplenic T cell Lymphoma
Received Date: Dec 27, 2022 / Accepted Date: Jan 07, 2023 / Published Date: Jan 27, 2023
Copyright: ©©2023: Anubha Bajaji. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Citation: Bajaji, A. (2023). Maraud and Gizzard-Hepatosplenic T cell Lymphoma. J Path Lab Med, 1(1), 07-10.
Abstract
Hepatosplenic T cell lymphoma is an exceptionally discerned, aggressive, extra-nodal lymphoma engendered from non activated, cytotoxic T lymphocytes which pre-eminently exemplify T cell receptor gamma/delta (TCRγδ). Hepatic parenchyma and bone marrow delineates a sinusoidal pattern of malignant T cell infiltration. Besides, quantifiable clonal T cells and abnormal T cells appear elevated within hepatic parenchyma, bone marrow, spleen or occasionally within peripheral blood.
Introduction
Hepatosplenic T cell lymphoma is an exceptionally discerned, aggressive, extra-nodal lymphoma engendered from non activated, cytotoxic T lymphocytes which pre-eminently exemplify T cell receptor gamma/delta (TCRγδ). Hepatic parenchyma and bone marrow delineates a sinusoidal pattern of malignant T cell infiltration. Besides, quantifiable clonal T cells and abnormal T cells appear elevated within hepatic parenchyma, bone marrow, spleen or occasionally within peripheral blood.
Previously designated as erythrophagocytic Tγ lymphoma, hepatosplenic T cell lymphoma is currently contemplated as a distinct entity. Surgical tissue sampling of hepatic parenchyma or bone marrow may be advantageous for cogent disease discernment. Typing of human leukocyte antigen (HLA) within incriminated subjects and siblings along with possible adoption of allogeneic stem cell transplantation is contemplated as a preliminary therapeutic manoeuver on account of rapid disease relapse upon discontinuation of therapy.
Hepatosplenic T cell lymphoma manifests < 1% of non Hodgkin’s lymphoma, configures ~2% of T cell / NK cell lymphomas and represents a T cell lymphoma which frequently incriminates the spleen.Factors contributing to emergence of hepatosplenic T cell lymphoma appear as male gender, accompanying history of inflammatory bowel disease, systemic immunosuppressive therapy, ongoing immunosuppression as encountered with solid organ transplant or bone marrow transplantation, Hodgkin’s lymphoma or infection with Plasmodium spp [1,2].
A male predominance is observed. The lymphoma commonly implicates young adult males with median age of disease emergence at ~35 years although paediatric subjects or elderly individualsmaymanifestthelymphoma.
Hepatosplenic T cell lymphoma demonstrates neoplastic clonal proliferation of gamma/delta (γδ) T cells. Cellular proliferation is induced by upregulation of JAK / STAT pathway composed of STAT3 and STAT5B along with PI3K signalling pathways. Alternatively, genetic mutations within chromatin modifiers as SETD2, INO80 and ARID1B may incur cellular proliferation [1,2].
Preponderantly (80%) of obscure aetiology, hepatosplenic T cell lymphoma may arise concurrent to chronic immunosuppression as encountered with renal or hepatic post-transplantation, immune-dysregulation as with inflammatory bowel disease, rheumatoid arthritis or administration of immunosuppressive agents as mercaptopurine, azathioprine or infliximabinanestimated20%instances.
Employment of tumour necrosis factor alpha (TNFα) inhibitors for treating conditions as Crohn's disease represents with significantly enhanced possible emergence of lymphomas as Bcellly mphomaorhepatosplenic Tcelllymphoma.
Hepatosplenic T cell lymphoma concurrent with inflammatory bowel disease is associated with contributory factors as thiopurine therapy of duration ≥ 2 years or disease emergence within male subjects. Besides, pertinent genetic predisposition and chronic antigenic stimulation may contribute to disease occurrence [1,2].
Hepatosplenic lymphoma of gamma/delta origin manifests bi-allelic rearrangement of TRG gene. Besides, chromatin modifying genes as SETD2, INO80, TET3 and SMARCA2 may be discerned within ~60% neoplasms. Genomic mutations confined to JAK / STAT pathway withmutationswithinSTAT3 and STAT5B may been countered.
Generally, genes such as RHOA, CD28 and CCR4 appear absent, in contrast to diverse T cell lymphomas. Methylation profiling of deoxyribonucleic acid (DNA)exhibits ~hyper-methylated genes as CD5, BCL11B, CXCR6, GIMAP7~hypo-methylated genes as ADARB1, NFIC, NR1H3, ST3GAL3. Besides, genomic mutation of PIK3CD may occur along with aberrant karyotype and isochromosome (7q) or ring chromosome 7.
Primarychromosomalaberrationsmanifestas~lossof7p22.1p14.1 ~gain of 7q22.11q31.1. Trisomy 8 may appear as a secondary event in ~70% instances. Initial representation of trisomy 8 is associated with significantly decimated overall survival and progression free survival (3,4).Majority (~80%) of incriminated subjects frequently exemplify hepatomegaly and splenomegaly. Massive splenomegaly is commonly encountered and is defined as spleen extending ≥ 6 cm below costal margin or spleen magnitude ≥ 20 cm upon greatest dimension or spleen weighing > 1000 grams upon splenectomy. Abdominal discomfort may ensure.
B clinical symptoms as pyrexia, night sweats and >10% loss of body weight may occur. Regional lymph node enlargement is uncommonly observed within < 25% subjects. Nevertheless, peripheral splenic lymph nodes may be incriminated [3,4]. Peripheral blood infrequently enunciates circulating lymphoma cells upon initial disease representation. Accompanying singular lineage or bi-lineage cellular decimation or cytopenias with pancytopenia is denominated by occurrence of moderate anaemia, thrombocytopenia and neutropenia.
Hyper-splenism, infiltration of bone marrow or disease manifestations concurrent to altered immunity are commonly observed. Lymphocytosis occurs within ~10% instances [3,4]. Bone marrow may be incriminated upon initial disease representation. Cutaneous surfaces exceptionally demonstrate disease involvement.
Cytological examination exhibits variable, cellular smears comprised of monotonous, miniature to intermediate cells incorporated with pale, a-granular cytoplasm, spherical to elliptical nuclei, moderately condensed nuclear chromatin and inconspicuous nucleoli [3,.4].
Cellular blasts appear as intermediate to enlarged cells with enhanced nucleo- cytoplasmic ratio, finely dispersed nuclear chromatin and absent nucleoli.
Incriminated bone marrow exhibits tri-lineage dyspoies is wherein dysmegakaryopoiesis is frequently discerned. Megakaryocytes depict hypo-lobated or miniature nuclei. Hemopha gocytosis may ensue in below<5%instances [3,4].
Upon gross examination, hepatic parenchyma and spleen manifest diffuse, homogeneous enlargement with absence of distinct, macroscopic lesions. Regional lymph node enlargement is absent. Upon microscopy, spleen demonstrates diffuse dissemination of lymphoma cells, cellular expansion within cords of red pulp and sinusoids along with atrophy or absence of white pulp.
Hemophagocytosis may be discerned [3,4]. Hepatic parenchyma and bone marrow delineate sinusoidal pattern of neoplastic infiltration. Bone marrow tumour burden appears concurrent to neutropenia. Median bone marrow tumour burden appears at ~30% and ranges from 5% to 80% [3,4].
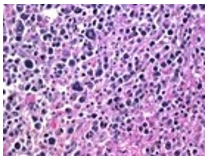
Figure 1 Hepatosplenic T cell lymphoma comprised of neoplastic cells demonstrating miniature to medium cells imbued with pale, a-granular cytoplasm, elliptical nuclei, condensed nuclear chromatin, inconspicuous nucleoli admixed with numerous blasts [4,5].
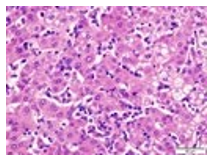
Figure 2 Hepatosplenic T cell lymphoma depicting miniature to intermediate neoplastic lymphocytes incorporated with pale cytoplasm with absent granules, elliptical nuclei, condensed chromatin and inconspicuous nucleoli [4,5].
Hepatosplenic T cell lymphoma is immune reactive to T cell antigens CD2, CD3, CD7, T cell intracellular antigen (TIA1), granzyme M, Fas ligand, CD56 and T cell receptor gamma/delta (TCRγδ). Ki67 proliferative index is variable.
Hepatosplenic T cell lymphoma is immune non reactive to CD1a, CD4, CD5, CD8, CD10, CD57, CD94, terminal deoxynucleotidyl transferase (TdT), Epstein Barr virus encoded small RNAs (EBER) or B cell antigens as CD19, CD20 and CD25. Besides, mature and non activated cytotoxic T cell immuno-phenotype appear non reactive to granzyme B and perforin. Also, T cell receptor alpha/beta (TCRαβ) and follicular T cell markers as PD-1, CD10, BCL6, CXCL13, ICOS appear immune non reactive. Exceptional instances are immune non reactive to T cell receptor gamma/delta(TCRγδ) and T cell receptor alpha/beta (TCRαβ) (TCR silent) [4,5].
Haematological or biochemical parameters as complete blood count (CBC), prothrombin time (PT)/international normalized ratio (INR), serum haptoglobin, blood urea nitrogen (BUN), serum creatinine, blood urea nitrogen/creatinine ratio, serum uric acid, lactic acid, serum albumin, alanine transaminase, aspartate transaminase, alkaline phosphatase, serum bilirubin, lactate dehydrogenase (LDH) and serum ferritin require evaluation [4,5].
Following confirmation of lymphoma, human leucocyte antigen (HLA) typing of incriminated subjects and siblings is recommended. Hepatosplenic T cell lymphoma manifests elevated serum lactate dehydrogenase (LDH), beta 2(β2) micro-globulin and bilirubin levels.
Upon flow cytometry, majority (80%) of hepatosplenic T cell lymphomas depict T cell receptor gamma/delta (TCRγδ) along with CD2+, CD3+, CD7+, CD56+ or KIR+. CD16 or CD94 is variably expressed and may be dim or absent. Neoplastic cells appear devoid of CD4-, CD5-, CD8- and CD57-.Immuno-phenotyping with flow cytometry of peripheral blood or bone marrow constituents depicts CD2+, CD3+, CD4-, CD5-, CD7+ and CD8- cellular phenotypes.
Besides, assessment of cellular beta F-1 (βF-1), gamma/delta and CD52 is recommended. Bone marrow or peripheral blood cytogenetics or fluorescent in situ hybridization (FISH) frequently exhibits isochromosome 7q and trisomy 8. Analysis of T cell receptor for clonal gamma/delta genetic rearrangement may be frequently adopted and appears diagnostic [4,5].
Hepatosplenic T cell lymphoma may be simulated by conditions such as acute viral hepatitis, leptospirosis, veno-occlusive disease, idiopathic thrombocytopenic purpura, hemophagocytic syndrome and benign or reactive augmentation of circulating gamma/delta T cells.
Hepatosplenic T cell lymphoma requires segregation from neoplasms such as T cell large granular lymphocytic (LGL) leukaemia, aggressive NK cell leukaemia / lymphoma, T cell prolymphocytic leukaemia, T cell lymphoblastic leukaemia / lymphoma, splenic marginal zone lymphoma, chronic active Epstein Barr viral infection, non splenic cytotoxic T cell lymphoma with γδ+ phenotype, myelo-proliferative disorders and infiltrative diseases as amyloidosis or sarcoidosis [4,5].
Ultrasonography is optimal for discerning hepatic and splenic architecture, magnitude and vascular outflow. Computerized tomography (CT) is beneficial for detecting absence of localized or regional lymphadenopathy.Computerized tomography (CT) or magnetic resonance imaging (MRI) exhibits homogenous hepatosplenomegaly. Fluorodeoxyglucose positron emission tomography (FDG PET / CT) represents diffuse uptake of fluorodeoxyglucose within incriminated areas as hepatic parenchyma, spleen or bone marrow [4,5].
Hepatosplenic T cell lymphoma may be appropriately treated with intravenous hydration, allopurinol and combination chemotherapy. Although standardized induction or chemotherapeutic guidelines for treating hepatosplenic T cell lymphoma are absent, cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) or CHOP-like therapies appear minimally efficacious in adequate disease eradication.
Nevertheless, chemotherapeutic agents as cyclophosphamide, doxorubicin, vincristine and prednisone (CHOP) or CHOP-like regimen, cyclophosphamide, vincristine sulfate, adriamycin and dexamethasone(hyper CVAD) or hyper CVAD-like regimen or non CHOP induction regimen comprised of 2' deoxycoformycin can be beneficially adopted to treat the lymphoma.Consolidative stem cell transplant (SCT) is optimal within subjects eligible for the therapeutic manoeuver.
Potential target therapy employing CD52, JAK1 / 2, STAT3, STAT5B and PI3KCD molecules appears advantageous Etoposide can be employed as a singular agent for treating hemophagocytic syndrome [4,5].
Besides, etoposide containing intense chemotherapeutic regimens such as ifosfamide, carboplatin, etoposide(ICE), ifosfamide, etoposide, high dose cytarabine(IVAC), etoposide, doxorubicin, vincristine, prednisone and cyclophosphamide(EPOCH) or etoposide, methylprednisolone, high dose cytarabine or Ara-C and cisplatin(ESHAP) may be advantageously utilized [4,5].
Regimens such as dihydroxyacetone phosphate (DHAP) and hyper CVAD appear advantageous in inducing disease remissions. However, singular conventional chemotherapies may be inadequate for disease alleviation and kinetic failures as neoplastic re-emergence between chemotherapeutic cycles may arise [4,5].
Consolidative therapy employing high dose chemotherapy and stem cell transplantation within first remission is recommended. Allogeneic stem cell transplantation is a preferential mode of therapy. Besides, long term disease remission may ensue with autologous stem cells in the absence of appropriate stem cell donor [4,5].
Along with intensive chemotherapy, antiviral agent as acyclovir, antifungal as fluconazole, anti pneumocystis pneumonia (PCP) drugs as trimethoprim/sulfamethoxazole may be adopted to treat the lymphoma. Frequent monitoring of complete blood count (CBC) is necessitated for assessing transfusion requirements. Central nervous system (CNS) prophylaxis remains as an obscure contingency [4,5].
Achemo-sensitive therapeutic response with significant decimation of tumour magnitude is common following initial treatment cycle. Besides, intense monitoring is necessitated for discerning chemo-refractory disease and employment of additional cycles of chemotherapy. Inadequate response to initial therapy or disease reoccurrence can be managed with drugs such as alemtuzumab, pentostatin or pralatrexate. Hepatosplenic T cell lymphoma is associated with an inferior prognostic outcome.
Majority of neoplasms are accompanied by disease relapse. Median progression free survival (PFS) appears at 9.5 months and median overall survival (OS) is ~12 months although overall survival ranges from 3 months to 34 months. Three year overall survival appears at an estimated ~37.6% whereas five year overall survival is around 31.6% [4,5].
Prognostic factors contributing to inferior outcomes manifest as~serum bilirubin > 1.5 milligram/decilitre~expression of T cell receptor alpha/beta(TCRαβ) ~trisomy 8.Preliminary disease recognition and implementation of high dose chemotherapy with stem cell transplantation while assessing limitations of CHOP therapy is optimal in achieving long term disease remission [4,5].
References
- Chowdhury, Z., Khonglah, Y. et al. (2022). Hepatosplenic T Cell Lymphoma: Diagnostic Conundrum. Int J Hematol Oncol Stem Cell Res. 16(1):66-73.
- Metelli, F., Solimando, R., Alemanni, L. V., Gafà, R., & Marasco, G. (2022). Hepatosplenic T-Cell Lymphoma Mimicking Acute Onset of Cholestatic Hepatitis in a Young Immunocompetent Man: A Case Report. Gastroenterology Insights, 13(3), 258-263.
- Honda, T., Yamaoka, M., Terao, Y. M., Hasegawa, D.,Kumamoto, T., Takagi, M., ... & Akiyama, M. (2022). Successful treatment of hepatosplenic T-cell lymphoma with fludarabine, high-dose cytarabine and subsequent unrelated umbilical cord blood transplantation. International Journal of Hematology, 115(1), 140-145.
- Pro, B., Allen, P., & Behdad, A. (2020). Hepatosplenic T-cell lymphoma: a rare but challenging entity. Blood, 136(18), 2018-2026.
- Huang, W., Xue, S., Zhang, Y., Liu, F., Tian, M., Wang, Y., ... & Wang, J. (2022). Refractory hepatosplenic T-cell lymphoma was successfully treated with salvage allogeneic hematopoietic stem cell transplantation combined with enhanced myeloablative preconditioning. Annals of Hematology, 1-6.