



Case Report - (2023) Volume 2, Issue 1
Idiopathic Bullous Pyoderma Gangrenosum in a 12-Year-Old Native American Male: A Case Report and Literature Review
Received Date: Mar 08, 2023 / Accepted Date: Apr 08, 2023 / Published Date: Jun 15, 2023
Copyright: ©Â©2023 Hunter A. Oâ??Connor, et al.,This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Citation: O
Abstract
Background: Pyoderma gangrenosum (PG) is a rare autoinflammatory skin disorder that presents with extremely painful ulcer- ative skin lesions. Bullous PG is a rare subtype, characterized by formation of large hemorrhagic bullae. The bullous subtype is especially uncommon in children. We present here a case of bullous PG in a young Native American male.
Case Presentation: A 12-year-old previously healthy Native American male initially developed several pustules on his back and axilla. Over the following three days, the lesions expanded into large hemorrhagic bullae. Suspecting an infectious etiology, he was initially treated with oral and topical antibiotics. The lesions continued to grow in size and number, and some developed into deep ulcerations. Antibiotic coverage was broadened, and he was admitted to a local hospital. The lesions progressed to include all four extremities, the torso, and deep ulcerations of the bilateral axillae. He was transferred to a higher-level facility for further workup. The diagnosis of bullous pyoderma gangrenosum was then made based on histologic findings of neutrophilic infiltration of the dermis and negative workup for other conditions. He was initially treated with systemic corticosteroids with immediate improvement. He continued to experience extreme pain, necessitating PICU admission for sedation during dressing changes. After three weeks, the lesions had healed significantly. He was given an infusion of Infliximab and discharged home. Two months after discharge, healing was nearly complete, and he was again infused with Infliximab. 4.5 months after discharge, his lesions had completely healed, albeit with significant scarring.
Conclusions: To our knowledge, less than 10 cases of pediatric bullous pyoderma gangrenosum have been reported. Addition- ally, we are not aware of any other reported cases of bullous PG occurring in a Native American child. PG is often associated with systemic diseases such as inflammatory bowel disease, and hematologic malignancy but no underlying systemic condition was ever found in the current case. As shown in this case, PG is often mistaken for infection, which can delay diagnosis and increase long term morbidity.
Keywords
Bullous Pyoderma Gangrenosum, Pediatric Autoimmunity, Idiopathic Pyoderma Gangrenosum, Neutrophilic Derma¬tosis, Ulcerative Skin DisorderList Of Abbreviations
CRP: C-reactive protein
ESR: Erythrocyte sedimentation rate
IBD: inflammatory bowel disease
IL: Interleukin IVIg: Intravenous immunoglobulin
JAK/STAT: Janus kinase and signal transducer and activator of transcription protein
NSAID: nonsteroidal autoinflammatory drug
PAPA: pyogenic arthritis, pyoderma gangrenosum, and acne syndrome
PAPASH: pyogenic arthritis, pyoderma gangrenosum, acne, and hidradenitis suppurativa syndrome
PG: Pyoderma gangrenosum
PSTPIP: Proline-serine-threonine phosphatase-interacting pro- tein
WBC: White blood cells
Background
Pyoderma Gangrenosum (PG) is a rare ulcerative autoinflamma¬tory skin disorder that affects 3-10 people per million worldwide [1]. First described in 1908 by a French dermatologist, Louis Brocq, it was originally thought to have an infectious etiolo¬gy [2]. The exact pathophysiology remains unknown, but it is now believed to involve an overactive neutrophil response [3, 4]. There are several subtypes, including a classic and bullous form. The classic subtype is most common and is characterized by the formation of papules or pustules that rapidly expand into extremely painful necrotic based ulcers [3]. The bullous form is rare and can be differentiated from the classic type in that large hemorrhagic bullae develop prior to ulceration [5]. PG most commonly presents in patients between the age of 20 and 50 and less than 5% of cases occur in children [6,7]. Furthermore, the bullous subtype is extremely rare in children, with very few cases currently reported in the literature. We present here the case of diffuse idiopathic bullous pyoderma gangrenosum in a 12-year-old Native American male. This case represented a sig-nificant diagnostic challenge for providers and should serve as a reminder for physicians to consider PG if a patient presents with a rapidly progressive necrotic skin condition.Case Presentation
A 12-year-old previously healthy Native American male pre-sented to his PCP with a 3-day history of multiple hemorrhagic bullae on his extremities and abdomen (symptom onset plus 3 days (S+3 days)). He first noticed small white pustules on his lower right back and left axilla. Overnight, the pustules grew significantly in size and progressed to hemorrhagic bullae. His general practitioner initially suspected an infectious etiology and treated the patient with oral and topical antibiotics. The next day (S+4 days), the lesions became increasingly painful and more numerous, which prompted the patient’s family to present to their local ED. The ED provider noted he had developed numer¬ous new pustules on his hands and torso and the bullae from the previous day had ruptured into ulcers with necrotic bases. There were also expanding areas of erythema surrounding the lesions. His initial laboratory workup demonstrated an elevated white blood cell count (WBC), C-reactive protein (CRP), and erythro¬cyte sedimentation rate (ESR). He remained afebrile. Suspect¬ing an infectious process that had not responded to the previous antibiotic treatment, antibiotic coverage was broadened. He was transferred to a higher care facility where he was admitted for further workup. Figure 1 and Figure 2 demonstrate lesions at the time of admission and one day after admission respectively (S+5 and S+6 days).

Figure 1: Skin lesions 5 days after symptom onset. A) Left axilla B) Right flank axilla C) Left arm D) Lower extremities
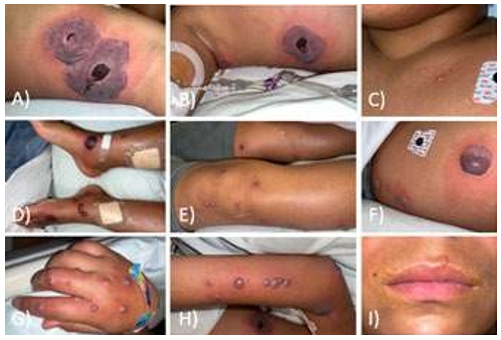
Figure 2: Skin lesions 6 days after symptom onset. A) Left axilla; B) Right flank and axilla; C) New lesions on left chest; D) and E) Lower extremities; F) Left flank; G) Right hand; H) Right arm; I) Upper lip lesion
On further questioning, he noted that the lesions had developed after swimming at a water park following a recent camping trip. The family reported that he may have initially had a bug bite on his right flank prior to symptom onset. He also had exposure to stray dogs that were known to carry lice and fleas. He also reported often playing in tall grass. He vaped but did not have any illicit drug use. He did not take any daily medications. He denied any weight loss, diarrhea, constipation, fevers, fatigue, or abdominal pain. His examination was benign except for the ulcers previously described.
On day two of admission (S+6 days), he developed high fevers for the first time. He continued to have elevated WBC and CRP/ ESR rose over the course of his hospital stay. Initial work-up involved a search for an infectious cause, included varicel-la-Zoster, tuberculosis, HIV, blood cultures for bacteria/fungal infection, acid-fast tissue stains, monkeypox, syphilis and an- ti-neutrophilic antibody screen, which were all negative. Initial¬ly, punch biopsy demonstrated mild spongiosis with few acute inflammatory cells and subepidermal splitting, but no histolog¬ic sign of infection was noted. He had severe pain throughout hospitalization requiring frequent doses of morphine and later a patient-controlled analgesia pump.
He continued to have high fevers, and his vital signs became un¬stable with recurrent hypotension. Antibiotics were continued, although no infectious source had been found. New lesions ap¬peared, and older lesions continued to spread and ulcerate, with eventual involvement of all four extremities, the torso, and full thickness ulcerations in bilateral axillae. The diagnosis remained unknown. On day five of admission (S+10 days), he was trans¬ferred again to a higher-level facility for specialist evaluation and further diagnostic workup. Figure 3 demonstrates the ulcers at time of discharge (S+10 days).

Figure 3: Skin lesions 10 days after symptom onset. A) Left axilla B) Right axilla C) Left arm; D) and E) Lower extremities; F) Right hand and arm
On admission to the outside facility, he was evaluated by dermatology, rheumatology, oncology, and infectious disease specialists. An extensive infectious workup, including a Karius study, was negative. An extensive autoimmune workup was also negative. Hematologic malignancy was ruled out based on normal peripheral smear and flow cytometry results, as well as an unremarkable CT abdomen pelvis. Repeat biopsy of the lesions demonstrated a dense dermal neutrophilic infiltrate, nearly extending into the subcutis, with subepidermal clefting. Based on these biopsy results, and an otherwise negative workup for other conditions, pyoderma gangrenosum was suspected. The patient was immediately started on IV corticosteroids (S+12 days), leading to noticeable improvement in the skin lesions after just three days (S+15 days). However, due to severe pain, the patient was transferred to the pediatric intensive care unit (PICU) for intensive pain control and sedation during dressing changes.
The patient remained in the PICU for the following four weeks (S+2 weeks to S+6 weeks). Intubation was required during the first 10 days due to severe pain and increasing sedation needs during dressing changes. Interestingly, Lyme IgM was found to be positive during this time, and he was treated with dox-ycycline. Through treatment with systemic corticosteroids and topical tacrolimus, the patient had progressive improvement of his skin lesions and pain. The corticosteroid dose was tapered down after two and half weeks of treatment. He was given an infusion of Infliximab, leading to further improvement of the skin lesions. One week after infliximab infusion, and five weeks after initial hospitalization (S+6 weeks), the patient was discharged home in stable condition.
The patient was discharged on an extended taper of oral corticosteroids and was instructed to follow-up for repeat Inflixamab infusions 2 and 6 weeks later. However, the patient was unable to follow-up for infliximab infusion until 8 weeks later (S+3.5 months). On examination at that time, he was noted to have diffuse 3 to 10 cm violaceous scars and two small active ulcers on the lower extremities. He was given another infusion of Inflixamab and was again scheduled for Inflixamab infusion 4 weeks later. However, he was again unable to present for the appointment and was not seen for another 2.5 months (S+6 months). The final examination at that time revealed complete healing, with no active lesions or pain. However, the patient had significant contracture scars of bilateral axilla that limit his range of motion as shown in Figure 4. At the time of writing this report, the patient is awaiting further evaluation to rule out inflammatory bowel disease and immunodeficiency, although suspicion for these conditions remains low.

Figure 4: Healed skin lesions 6 months after symptom onset. A) Left axilla and arm; B) Right axilla and arm C) Lower extremities;D) Left axilla with scar tear
Discussion
As this case demonstrates, PG is a challenging and urgent di-agnosis that many clinicians mistake for infection. PG usually occurs in middle aged adults, with an average age of presenta- tion around 51 [8]. PG is rare in children, accounting for only 4% of all cases [3]. There are several sub-classifications of PG, including classic/ulcerative, pustular, bullous/atypical, and veg¬etative variants [9]. The case described here represents the bul- lous subtype, which is characterized by rapidly evolving painful vesicles that enlarge into hemorrhagic bullae [9]. To our knowl¬edge, eight cases of pediatric bullous pyoderma gangrenosum have been reported since 1994. A recent systematic review of pediatric PG cases found that only seven cases of pediatric bul¬lous PG were reported between 1994 and 2016 and we are aware of only one additional case since 2016 [7,10]. Additionally, we are not aware of any other reported cases of pediatric bullous PG occurring in a Native American patient.
Association with Other Diseases
To date, no other disease was found to be associated with the case above. However, greater than 50% of PG cases in pediatric patients are associated with systemic disease [7]. Inflammatory bowel disease (IBD), equally split between Crohn’s and ulcer¬ative colitis, is most frequently associated with PG in children [7]. Other commonly associated conditions in children include hematologic malignancies, immune deficiencies, vasculitis, and PAPA syndrome [7]. The bullous subtype is most often idiopath¬ic in pediatric patients, but at least one case has been associated with Crohn’s disease, one with myelodysplastic syndrome and one with acute myeloid leukemia [7,10]. In the adult population, bullous PG is highly associated with hematologic malignancy, which is present in greater than 70% of cases [9, 11].
Pathogenesis
PG can be thought of as a neutrophilic dermatosis because pa-thology exhibits significant neutrophilic infiltration of the der-mis without evidence of infection [2]. The pathogenesis of PG is not fully understood but is currently thought to involve overacti-vation of neutrophils due to intrinsic neutrophil dysfunction, and abnormal production of inflammatory cytokines [12]. Recent studies have shown that PG neutrophils are dysfunctional in a variety of domains - abnormal chemotaxis, migration, phagocy-tosis, hyperactivity, and bactericidal ability [2, 3]. Studies have also found that several inflammatory cytokines are overproduced in PG lesions, including IL-8, and IL-1β [13]. IL-8 is a known neutrophil chemokine, and exposure of human skin xenografts to high levels of IL-8 has been shown to cause formation of PG like lesions [14]. The association of PG with other inflammatory diseases and improvement of the disease with agents that inhibit neutrophil function such as dapsone, and colchicine also support an etiology of underlying neutrophil dysfunction [2]. Interest¬ingly, there are many reports of PG occurring in patients with leukocyte adhesion deficiency type 1, a disorder wherein neu¬trophils are unable to extravasate into surrounding tissue [12, 7]. This finding suggests that development of PG is likely not solely due to neutrophils [12].
Genetic Syndromes
Several genetic syndromes and individual genetic mutations are associated with the development of PG. PAPA syndrome is char¬acterized by the triad of pyogenic sterile arthritis, cystic acne, and PG [12, 15]. This syndrome has been found to originate from a mutation in the PSTPIP-1 gene, which leads to increased production of the inflammatory cytokines IL-1β, and IL-16 [1]. A different mutation of the same gene results in PAPASH syn¬drome – characterized by a tetrad of pyogenic sterile arthritis, PG, cystic acne, and hidradenitis suppurativa [12, 15]. These syndromes follow an autosomal dominant inheritance pattern [15]. Although these conditions cannot be ruled out without of¬ficial genetic testing, they are unlikely in patients, such as the patient presented above, who do not have a family history of these conditions. PG has also been found to share several gene mutations with IBD - including IL-8RA, and tissue inhibitor of metalloproteinase 3 [12, 15]. Additionally, in patients with asso¬ciated polycythemia vera, mutations in the JAK/STAT pathway are thought to contribute to development of PG. [2, 4, 12].
Clinical Presentation
The clinical presentation of bullous PG involves formation of pustules or vesicles, that rapidly expand into hemorrhagic bullae that eventually develop into painful ulcerations. Lesions are usu¬ally associated with severe pain, which is often out of proportion to ulcer size [3]. Adults usually have less than three ulcers with the classic form preferentially affecting the lower extremities, and the bullous form affecting the upper extremities [3,6,8]. In contrast, children most commonly exhibit widespread disease, with ulcers affecting multiple anatomical locations [7]. PG lesions often demonstrate pathergy, with formation after minor trauma to the skin [8]. There have been many reports of the disease presenting or worsening after surgery due to pathergy [8]. Systemic symptoms such as fever, joint pain, malaise, and myal-gia are also often present [3]. The case presented above followed a relatively classic presentation for bullous PG in children. Of note, the patient developed wide-spread ulcerations that were extremely painful. The deep severe ulceration in the axilla may have been due to pathergy, as this is an area exposed to significant friction and disturbance with arm movement. Additionally, the patient had intermittent fevers that made it especially difficult to differentiate PG from an infectious etiology.
The presentation of PG mimics that of several other more com¬mon diseases, which often delays diagnosis. The differential di¬agnosis often includes infection of any cause, insect bites, sweet syndrome, systemic lupus erythematosus, granulomatosis with polyangiitis, cutaneous lymphoma, and Behcet disease, to name a few [4]. The median time from symptom onset to diagnosis of PG in pediatric patients has been reported to be around 2 months [7]. With a diagnosis made just 12 days after symptom onset, the PG case presented here was identified much quicker than most cases reported in the literature. This may be due to the severity of the presentation in this case.
Diagnosis
The diagnosis of PG is made based on the clinical manifesta-tions, and progression, along with histologic findings, and exclu-sion of other disease processes. In 2018, a study by Maverakis et al. set out to determine a set of diagnostic criteria to help phy¬sicians identify this disease [4]. The current consensus is that a biopsy demonstrating neutrophilic invasion into the dermis is a major criterion [4]. In addition, 4 out of 8 minor criteria must be present: histological exclusion of infection, pathergy, history of IBD, papules or pustules that rapidly ulcerates, multiple ulcers with at least one in the lower extremities, cribriform scars at healed sites, erythematous ulcers with undermining borders, and a decrease in ulcer size following immune suppression thera¬py [4]. Additionally, to determine the PG classification subtype, two separate minor and major criteria must be met [11]. For bul¬lous PG, the major criteria include painful inflammatory bullae that rapidly enlarge and coalesce with the exclusion of any other cause of bullae formation [11]. The minor criteria include pa-thology demonstrating neutrophils in the subepidermal bullae with or without necrosis, associated hematological malignancy, pathergy, and a positive response to steroids [11].
Treatment
Management of PG involves inflammation suppressing medi-cations, wound care, pain management, and identification and treatment of any underlying systemic conditions [1]. Small, localized lesions can often be managed with locally acting im-mune suppression agents such as topical corticosteroids, or topi-cal tacrolimus [1]. Diffuse, rapidly progressive PG is most often treated with systemic corticosteroids, cyclosporine, or biologic immunomodulators [1]. As reported in the case above, patients generally have rapid improvement when treated with these agents [8]. However, wounds that are refractory to traditional treatments can be treated with IVIg or alkylating agents [1]. Due to a lack of large randomized controlled trials on the treatment of PG, the optimum pharmacotherapy is currently unknown. Prop¬er wound care is required to prevent superinfection and promote healing. This usually involves gentle cleaning of ulcers with nor¬mal saline. Harsh cleansing or dressing techniques such as the use of wet to dry dressings or silver nitrate should be avoided as these may lead to pathergy [1]. Because the disease is so often associated with severe pain, opiates, NSAIDs, and other pain medications are often needed. With the widespread ulcerations in the case above, pain control was especially challenging, and even necessitated long term sedation. It is crucial to identify and promptly treat any underlying conditions, as some associated conditions, such as hematologic malignancy, and immunodefi¬ciency, have high mortality if not managed appropriately [7]. PG may be the first sign of one of these underlying conditions, so prompt investigation is imperative [7, 10].
Long Term Prognosis
The long-term outcomes of PG have not been well studied, but it appears that most cases resolve over time with appropriate treat-ment. Based on a few small case series, around 50 to 70% of cases will have complete healing within 6 months [5, 16], and up to 95% of cases will resolve within 3 years [5]. The patient in the case presented here had complete healing at 6-month fol¬low-up. However, as shown in Figure 4, healing of PG lesions often results in significant scarring and deformity. Recurrence of disease is common and can occur in 45 to 55% of cases which necessitates long term treatment [17]. Interestingly, recurrence of PG does not usually align with progression of the underlying systemic condition [9]. PG can rarely lead to death due to severe infection of diffuse lesions [17,18]. Mortality of patients with PG, especially pediatric patients, are most often due to under-lying hematologic malignancy, immunodeficiency, or vasculitis [7, 17, 19].
Conclusion and Main Learning Points
Pyoderma gangrenosum (PG) is a rare autoinflammatory skin disorder that presents with extremely painful, rapidly progres-sive ulcerations of the skin. PG rarely occurs in children, and the bullous subtype is especially uncommon in the pediatric popu-lation. PG in children is often associated with systemic diseases such as inflammatory bowel disease, hematologic malignancy, immune deficiency, and vasculitis. The pathogenesis of pyoder¬ma gangrenosum remains unknown but is likely related to an overactive neutrophil response and dysfunction of the innate im¬mune system.
Diagnosis is made based on clinical and histological features with exclusion of other disease processes. The disease is often mistaken for infection, which can delay diagnosis. Treatment in¬volves suppression of inflammation, usually with systemic corti¬costeroids, as well as wound care, pain control, and investigation and treatment of underlying conditions. High clinical suspicion, and prompt initiation of treatment is required as the disease can lead to significant long-term morbidity, and even mortality due to underlying conditions.
References
- Ahronowitz, I., Harp, J., & Shinkai, K. (2012). Etiology and management of pyoderma gangrenosum: a comprehensive review. American journal of clinical dermatology, 13, 191-211.
- Barbe, M., Batra, A., Golding, S., Hammond, O., Higgins, J. C., O’Connor, A., & Vlahovic, T. C. (2021). Pyoderma gangrenosum: a literature review. Clinics in Podiatric Medicine and Surgery, 38(4), 577-588.
- Gameiro, A., Pereira, N., Cardoso, J. C., & Gonçalo, M. (2015). Pyoderma gangrenosum: challenges and solutions. Clinical, cosmetic and investigational dermatology, 285-293.
- Maverakis, E., Ma, C., Shinkai, K., Fiorentino, D., Callen,J. P., Wollina, U., ... & Cheng, M. Y. (2018). Diagnostic criteria of ulcerative pyoderma gangrenosum: a Delphi consensus of international experts. JAMA dermatology, 154(4), 461-466.
- Bennett, M. L., Jackson, J. M., Jorizzo, J. L., Fleischer Jr, A. B., White, W. L., & Callen, J. P. (2000). Pyoderma gangrenosum a comparison of typical and atypical forms with an emphasis on time to remission. Case review of 86 patients from 2 institutions. Medicine, 79(1), 37-46.
- Langan, S. M., Groves, R. W., Card, T. R., & Gulliford,M. C. (2012). Incidence, mortality, and disease associations of pyoderma gangrenosum in the United Kingdom: a retrospective cohort study. Journal of investigative dermatology, 132(9), 2166-2170.
- Kechichian, E., Haber, R., Mourad, N., El Khoury, R., Jab-bour, S., & Tomb, R. (2017). Pediatric pyoderma gangrenosum: a systematic review and update. International journal of dermatology, 56(5), 486-495.
- Binus, A. M., Qureshi, A. A., Li, V. W., & Winterfield, L. S. (2011). Pyoderma gangrenosum: a retrospective review of patient characteristics, comorbidities and therapy in 103 patients. British Journal of Dermatology, 165(6), 1244-1250.
- Ruocco, E., Sangiuliano, S., Gravina, A. G., Miranda, A., & Nicoletti, G. (2009). Pyoderma gangrenosum: an updated review. Journal of the European Academy of Dermatology and Venereology, 23(9), 1008-1017.
- Arikan, K., Özsürekçi, Y., & Ceyhan, M. (2017). Bullous pyoderma gangrenosum as the presenting sign of acute myeloid leukemia in a child. Journal of pediatric hematology/oncology, 39(4), 312-313.
- Su, W. D., Davis, M. D., Weenig, R. H., Powell, F. C., & Perry, H. O. (2004). Pyoderma gangrenosum: clinicopatho-logic correlation and proposed diagnostic criteria. International journal of dermatology, 43(11), 790-800.
- Braswell, S. F., Kostopoulos, T. C., & Ortega-Loayza, A. G. (2015). Pathophysiology of pyoderma gangrenosum (PG): an updated review. Journal of the American Academy of Dermatology, 73(4), 691-698.
- Ortegaâ?Loayza, A. G., Nugent, W. H., Lucero, O. M., Washington, S. L., Nunley, J. R., & Walsh, S. W. (2018). Dys-regulation of inflammatory gene expression in lesional and nonlesional skin of patients with pyoderma gangrenosum. British Journal of Dermatology, 178(1), e35-e36.
- Oka, M., Berking, C., Nesbit, M., Satyamoorthy, K., Schaid-er, H., Murphy, G., ... & Herlyn, M. (2000). Interleukin-8 overexpression is present in pyoderma gangrenosum ulcers and leads to ulcer formation in human skin xenografts. Laboratory investigation, 80(4), 595-604.
- DeFilippis, E. M., Feldman, S. R., & Huang, W. W. (2015).The genetics of pyoderma gangrenosum and implications for treatment: a systematic review. British Journal of Dermatology, 172(6), 1487-1497.
- Saracino, A., Kelly, R., Liew, D., & Chong, A. (2011). Pyoderma gangrenosum requiring inpatient management: a report of 26 cases with follow up. Australasian journal of dermatology, 52(3), 218-221.
- Von den Driesch, P. (1997). Pyoderma gangrenosum: a report of 44 cases with followâ?up. British Journal of Dermatology, 137(6), 1000-1005.
- Mlika, R. B., Riahi, I., Fenniche, S., Mokni, M., Raouf Dhaoui, M., Dess, N., ... & Mokhtar, I. (2002). Pyoderma gangrenosum: a report of 21 cases. International journal of dermatology, 41(2), 65-68.
- Kaffenberger, B. H., Hinton, A., & Krishna, S. G. (2018). The impact of underlying disease state on outcomes in patients with pyoderma gangrenosum: A national survey. Journal of the American Academy of Dermatology, 79(4), 659-663.