
International Journal of Natural Sciences and Interdisciplinary Research(IJNSIR)
ISSN: 3143-1046 | DOI: 10.33140/IJNSIR


Research Article - (2026) Volume 1, Issue 1
Cyclopentadiene is not Aromatic and not Planar Like Benzene: Yet Deprotonation (pKa) from its Lone sp3 Carbon Obeys Linear Free Energy Relationships (LFER)
2Department of Chemistry, Osmania University, India
3Department of Pharmacy, School of Health Science, University of KwaZulu-Natal, Durban, South Africa
Received Date: Mar 09, 2026 / Accepted Date: Apr 03, 2026 / Published Date: Apr 15, 2026
Copyright: ©2026 V. Jagannadham, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Citation: Jagannadham, V., Rachuru, S., Padmavathi, D. A., Skelton, A. A. (2026). Cyclopentadiene is not Aromatic and not Planar Like Benzene: Yet Deprotonation (pKa) from its Lone sp3 Carbon Obeys Linear Free Energy Relationships (LFER). Int Nat Sci Int Rese, 1(1), 01-07.
Abstract
Cyclopentadiene (CP) and its seven Cyano substituted derivatives obeyed Linear Free Energy Relationship (LFER) with an excellent correlation for pKa s of deprotonation equilibrium from the lone sp3 carbon with Hammett σm values for the substituents at 2 and 3 positions, Taft σ* values for 1 and 4 positions of CP; for disubstituted derivatives at 1or 4 and 2 or 3 positions we applied the sum of Taft σ* and Hammett σm values respectively. Deprotonation is facilitated by electron withdrawing capacity of the Cyano group with a positive Hammett-Taft reaction constant (ρHT ; HT stands for Hammett-Taft) of 2.75. To the best of our knowledge, we are the first ones to apply the summation of Hammett and Taft σ*. Further, we determined the pKa of the deprotonation equilibrium of unknown 1,4-dicyanoCP using the Hammett-Taft plot; the pKa of this compound in question was also determined theoretically.
Keywords
Cyclopentadiene, LFER, Hammett Equation, Taft Equation, DFT and pKa
Introduction
We have recently reported the application of LFER to the deprotonation equilibriums of N(1)-H acidities of five membered nitrogen heterocycles as a small review and isoxazolium cations as a small note; several references were cited in both the publications [1,2]. In these studies, a lucid and clear identification of substituents has arrived in terms of their ortho, meta and para positions using a visual comparison with benzene [2]. Recently the study on substituent effects was reported by Yongge Qiu in the Diels-Alder reaction of CP and maleic anhydride [3]. He used Hammett σp substituent constants for both 1- and 2- substituted CPs without giving any justification. In the present work, though cyclopentadiene is not aromatic and not planar like benzene, a successful application of LFER was observed. Suitable explanations are given.
Methods
Linear correlation was done using the KaleidaGraph software, Version 4.1, Reading, PA, USA. The chemical structures are drawn using ChemDraw software. The pKa values of all cyclopentadiene and its cyano substituted derivatives used in this work are from reference [4]. Gaussian 09 program [5] was used for all quantum mechanical calculations [5]. The pKa values were determined using SMDsSAS (scaled solvent-accessible surface) model and the geometries were optimized at the M06-2X [6] /6-31+(d,p) level like Lian et. al [7]. The nonspecific form of equation 1 of Scheme 1 is given by Equation 2 [7]:
![]()

In the above equation Go (H+) = –6.29 kcal mol-1 and the experimentally measured hydration free energy ΔG* aq,solv (H+) = –265.9 kcal mol-1 was taken from the literature [8-13]. ΔGo→* is the gas-phase standard correction and is used to convert from 1 atm ideal gas standard state to 1 M standard state, where superscripts o and * indicate 1 atm and 1 M standard states, respectively: ΔGo→* = RT ln 24.26 = 1.89 kcal mol–1 at 298 K [12]. The estimation of the value of Go (H+) = –6.29 kcal mol-1 is from reference [8].
Discussion
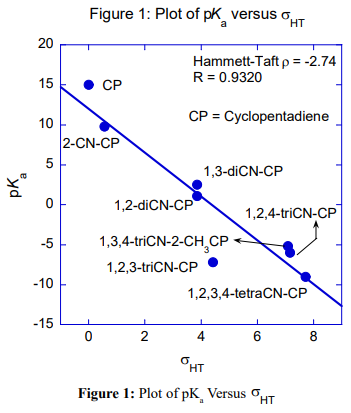
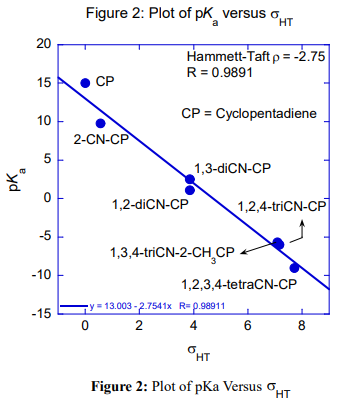
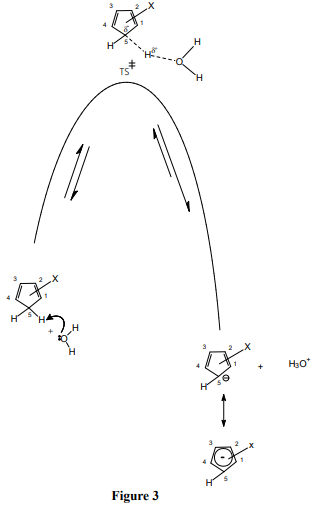
Table 1 is the pKa data of CPs. Figures 1 and 2 are the Hammett-Taft plots of pKa versus σHT (here HT stands for Hammett-Taft). The dissociation equilibrium of cyclopentadiene is shown in scheme 1. As shown in our earlier publication [2] using visual comparison, the 2 and 3 positions are assumed to be meta to the deprotonation site from the lone sp3 carbon of cyclopentadiene [2]. Positions 1 and 4 are assumed to be ortho to the deprotonation site. Accordingly, the substituent constants are summed up using Hammett and Taft substituent constants; these are clearly shown in column 3 (bold), Table 1. With the assumption described in the preceding paragraph, LFER plots are drawn; pKa versus σHT are shown in figures 1 and 2. The loci of the plots are with negative sign. But they are to be understood positive values as log Ka would have been plotted against HT. The positive values as log Ka would have been plotted against HT. The LFER positive reaction constant ( HT) indicates that the electron withdrawing CN substituents makes the cyclopentadiene more and more acidic. A striking question we may anticipate from the readers is that Hammett equation originated from the study of ionization of meta and para substituted benzoic acids which are typically aromatic.And Taft equation originated from the study of acid and base catalyzed hydrolysis of ethyl acetates and then correcting the σ* values by a factor of 2.48 [14-17]. The factor 2.48 is the mean Hammett value of acid and base catalyzed hydrolysis of meta and para substituted methyl and ethyl benzoates and again which are typically aromatic. The substituent effects are transmitted efficiently to the reaction site through the delocalized π-electron cloud in the benzene ring. And they successfully follow LFER. The present study deals with the ionization of CP to give cyclopentadienyl anion. CP is not aromatic and not planar like benzene. It has no uninterrupted cloud of π-electrons. Yet, its deprotonation equilibrium is obeying LFER. The conjugate acid, CP, has a pKa of 15 and is abnormally strongly acidic hydrocarbon by a driving force of aromatization to yield stable and planar cyclopentadienyl anion. It has an uninterrupted cloud of π-electrons which can transmit substituent effect to the ionization site from the substituent in the five membered CP ring. Figure 1 is the LFER plot wherein we have taken Taft σ* values for 1, 4 positions and Hammett σm values for 2 and 3 positions. Though the correlation coefficient is little poor (R = 0.9314) the trend is unmistakable. A replot of figure 1 is given in figure 2 without the point for 1,2,3-cyanoCP. The correlation is excellent with a correlation coefficient (R) of 0.9891 close to unity. At this juncture of research, we are not able to explain the deviation of the point of 1,2,3-cyanoCP. Though the CP is not aromatic the explanation for this is that the cyclopentadienyl anion is a planar, cyclic, regular-pentagonal ion; it has 6 π-electrons (4n + 2, where n = 1), which satisfies Hückel's rule of aromaticity. Therefore, cyclopentadiene aromatic anion with delocalized π electrons is more stable. The structure shown in scheme 1 with a circle inside the 5 membered pentagon is a resultant of five resonance structures in which each carbon atom carries about 1/5th of the total negative charge. Even the sodium and potassium salts of cyclopentadienyl anion, sodium/potassium CPs are known to be stable salts [18,19]. The fact that figure 2 conforms to Hammett’s LFER, it does reflect that 1,2 and 3,4 are ortho and meta positions respectively. There was a very detailed report about the thermodynamics of ionization of cyano carbon acids [20]. It was concluded in that study that the overall negative entropy is a favorable result for the ionization of cyano carbon acids. Therefore, though we have not performed any thermodynamic study in the present work, there is no reason for not assuming that the transition state for the deprotonation process may look more like the product type than the reactant type as shown in figure 3. Hence, the developing partial negative charge on the lone sp3 carbon of the transition state makes the reactant approach planarity and aromaticity making the species transmit substituent effects efficiently. Therefore, the deprotonation process of CP conforms to LFER with a major contribution of the substituent effects from the product like transition state. Figure 2 enabled us to determine the pKa of unknown compound 1,4-diCyanoCP. To the best of our knowledge the pKa of this compound is not reported in the literature. By taking Taft σ* value as 3.3 + 3.3 = 6.6 and substituting this value in the straight-line equation of Figure 2 i.e., y =13.003 -2.7541x, we get the pKa of 1,4-diCynoCP as -5.18. Theoretical determination 1,4-diCyanoCP turned out to be -4.52. We have determined pKa’s of all the compounds by adopting SMDsSAS model [7]. Since we did not get the pKa anywhere close to -5.17 (pKa of 1,4-diCP) at M06-2X/6-31+G(d,p) level [7] the geometry of 1,4-diCP was optimized at M06-2X/6-311++G(d,p) level. This level has a third layer of valence functions composed of uncontracted primitive set and good for final accurate measurements of energies (thoughcomputationally more expensive); hence we have used this basis set. We have determined pKa’s of all the compounds used by adopting SMDsSAS model [7]. The crucial α value for the SMDsSAS model and their corresponding free energies are given in Table 2. The fact that we are getting almost the same value (pKa of 1,4-diCynoCP) by both graphical LFER and theoretical method, it does reflect that our theoretical determination of pKa value is correct and bolster our visualization of 1,2 position as ortho to the deprotonation site.
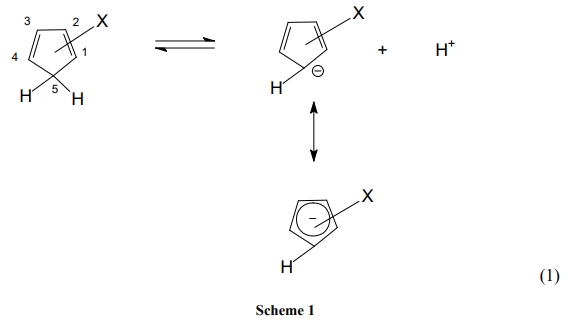
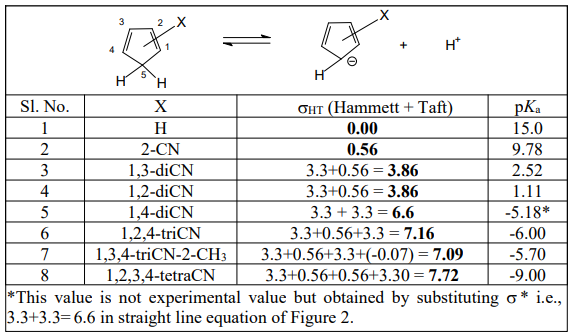
Table 1: pKa Data of Cyclopentadienes
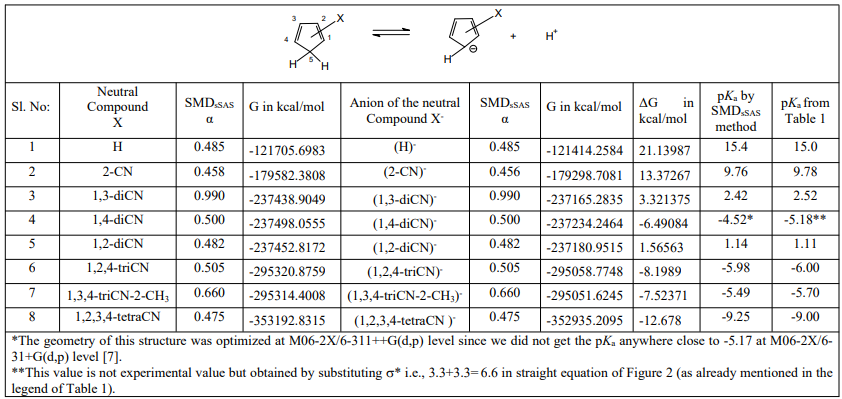
Table 2: Theoretical pKa Data of Cyclopentadienes Determined by SMDsSAS Model. The Geometries of all the Structures were Optimized at M06-2X/6-31+G(d,p) level [6]
Conclusion
CP is non-aromatic, but its deprotonation conforms to LFER. The primary plausible reason for this is that the TS resembles the aromatic product that is its corresponding anion. The fact that we are getting pKa value for 1,4-dicynoCP (which is not reported in the literature) determined by both theoretical (-4.52) and by Hammett-Taft LFER (-5.18) plot almost the same, it does reflect the value is reasonably correct; Thus, a pertinent and important inference from the preceding statement is that this also bolsters our visualization of 1, 4 position in Cyclopentadiene correspond to ortho relative to deprotonation site. The good linearity of Hammett-Taft plot (Figure 2) clearly reflects that 1,4 and 2,3 correspond to ortho and meta positions respectively. What is not comprehensible is that Yongge Qiu [3] in his article has taken para position in Cyclopentadiene and had used Hammett σp substituent constants; the discussion in our earlier article [1] and the findings in our present article clearly reflect that para position does not exist [1,3].
Acknowledgment: The authors are grateful to the Centre for High Performance Computing (CHPC), Cape Town, South Africa, for their generous allocation of supercomputer time.
Funding
The research did not receive any specific funding.
Conflict of interest statement
The authors declare no conflicts of interest.
Availability of data and material
The supplementary file consists of all the required computational input and output files.
Authors Statement of contributions
All the authors make equal contribution.
References
- Rachuru, S., Jagannadham,V. (2022). Application of LFER to the N(1)-H Acidities of Five-Membered Nitrogen Heterocyclic Ring Systems: A Review on Graduate Chemical Education Exercise. Current Physical Chemistry (Bentham Science Publishers), vol. 12, page 117-127.
- Rachuru, S., Manthena, R., Ankamma, K., Padmavathi, D. A., Jagannadham, V.(2024). Application of Hammett and Taft Equations Together on the Deprotonation Equilibriums of Isoxazolium Cations: A One Hour Graduate Classroom Teaching. World Journal of Chemical Education, vol. 12(1),1-5.
- Yongge, Q. (2015). Substituent effects in the Diels–Alder reactions of butadienes, cyclopentadienes, furans and pyroles with maleic anhydride. J. Phys. Org. Chem, 28 370–376.
- Webster, O. W. (1966). Polycyanation. The Reaction of Cyanogen Chloride, Cyclopentadiene, and Sodium Hydride. J. Am. Chem. Soc., 88, page 3046.
- Zhao, Y., Truhlar, D. G. (2008). The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor Chem Account 120, 215–241.
- Gaussian 09, Revision E.01, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. et al. (2013). Solvent Effects on the [3+2] Cycloaddition of 2-Furfural Oxime and Ethyl Propiolate: Unexpected Change in Regioselectivity. Science and Education An Open Access and Academic Publisher. Vol 5(1), 6-10.
- Lian, P., Johnston, R. C., Parks, J. M., Smith, J. C. (2018). Quantum Chemical Calculation of pKas of Environmentally Relevant Functional Groups: Carboxylic Acids, Amines, and Thiols in Aqueous Solution. J. Phys. Chem. A 2018, 122, 4366.
- Thapa, B., Schlegel, H. B. (2016). Density Functional Theory Calculation of pKa’s of Thiols in Aqueous Solution Using Explicit Water Molecules and the Polarizable Continuum Model. J. Phys. Chem. A. 120, 5726.
- Camaioni, D. M., Schwerdtfeger, C. A. (2005). Comment on “Accurate Experimental Values for the Free Energies of Hydration of H+, OH-, and H3O+”. J. Phys. Chem. A, 109, 10795. 10795.
- Isse, A. A., Gennaro, A. (2010). Absolute Potential of the Standard Hydrogen Electrode and the Problem of Interconversion of Potentials in Different Solvents. J. Phys. Chem. B, 114, 7894.
- Kelly, C. P., Cramer, C. J., Truhlar, D. G. (2006). Aqueous Solvation Free Energies of Ions and Ion−Water Clusters Based on an Accurate Value for the Absolute Aqueous Solvation Free Energy of the Proton. J. Phys. Chem. B, 110, 16066.
- Marenich, A. V., Ho, J., Coote, M. L., Cramer, C. J., Truhlar,D. G. (2014). Computational electrochemistry: prediction of liquid-phase reduction potentials. Physical. Chem. Chemical Phys, 16, 15068.
- Marenich, A. V., Cramer, C. J., Truhlar, D. G. (2009). Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B, 113, 6378.
- Jagannadham, V., Sanjeev, R. (2012). The Significance and the Basis for the Taft ρ* Value: A One Hour Physical-Organic Chemistry Classroom Lecture to Graduate Students. Physical Chemistry, Published by Scientific and Academic Publishing, USA, (2012) vol. 2 page 100-102.
- Ingold, C. K. (1930). “Mechanisms of acid and base catalyzed hydrolysis of esters”, J. Chem. Soc, page 1032.
- Neil S. Issacs. (1987). “Physical Organic Chemistry”, ELBS/ Longman, page 154 and 297.
- Colin D. Johnson. (1980). “The Hammett equation”, (Cambridge Texts in Chemistry and Biochemistry), page 75.
- Cyclopentadienide. (2016). PubChem Compound Database. National Center for Biotechnology Information.
- Panda, T. K., Gamer, M. T., Roesky, P.W. (2003). "An Improved Synthesis of Sodium and Potassium Cyclopentadienide" Organometallics, 22, 877–878.
- Boyd, R. H., Wang, Chin-Hsien. (1965). The Thermodynamics of Ionization of Cyanocarbon Acids. J. Am. Chem. Soc., 87, page 430-436.