
International Journal of Clinical and Medical Education Research(IJCMER)
ISSN: 2832-7705 | DOI: 10.33140/IJCMER
Impact Factor: 0.93


Research Article - (2025) Volume 4, Issue 4
Computational Identification of Potential Ligands for SARS-CoV-2 Spike Protein: A Docking Study with Natural Compounds and Antiviral Drugs
Received Date: May 29, 2025 / Accepted Date: Jun 26, 2025 / Published Date: Jul 02, 2025
Copyright: ©Â©2025 Alessandro Careglio. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Citation: Careglio, A. (2025). Computational Identification of Potential Ligands for SARS-CoV-2 Spike Protein: A Docking Study with Natural Compounds and Antiviral Drugs. Int J Clin Med Edu Res, 4(4), 01-13.
Abstract
The ongoing global challenge posed by SARS-CoV-2 highlights the urgent need for novel antiviral strategies, particularly targeting the viral Spike protein crucial for host cell entry. This computational study aimed to identify potential ligands for the SARS-CoV-2 Spike protein using molecular docking simulations. Both conventional and covalent docking approaches were employed to screen a diverse set of compounds, including natural products from various plant matrices (cocoa, Hypericum perforatum, Hedera helix, wormwood, sage) and approved or experimental antiviral drugs, with a specific focus on nitrile-containing compounds for covalent interactions with Cysteine 136. For conventional docking, several natural compounds demonstrated high binding affinities. Notably, epicatechin gallate and procyanidin A2 from cocoa, and compounds from Hypericum perforatum (e.g., hypericin, amentoflavone, biapigenin) showed promising scores and favorable ADME profiles. Ivy compounds like hederacoside and alpha-hederin exhibited high scores and were observed to occupy key amino acids within the Spike protein's ACE2 binding interface. Among the antiviral drugs, Ritonavir yielded some of the best docking scores, particularly with the receptor-bound Spike conformation, while Oolonghomobisflavan-A and -B also displayed excellent scores and are known to inhibit COVID proteases.
In covalent docking targeting Cysteine 136, Bauhinin and Bursatellin emerged as top-scoring natural compounds. Among drugs, the beta-blocker Epanolol and the antidepressant Vilazodone showed significant binding to this exposed cysteine residue. This finding for Vilazodone is particularly notable given existing clinical studies suggesting an association between antidepressant use, including Vilazodone, and reduced risk of severe COVID-19 outcomes. In conclusion, this study identifies several promising natural compounds and existing drugs as potential Spike protein ligands through both conventional and covalent binding mechanisms. These findings warrant further experimental validation, potentially exploring the synergistic effects of natural compound mixtures, and investigating the clinical implications of identified drug-protein interactions.
Keywords
SARS-CoV-2, Spike Protein, Molecular Docking, Natural Compounds (Phytocompounds), Covalent Docking
List Of Abbreviations
C : Catechin
EC : Epicatechin
ECG : Epicatechin gallate
EGC : Epigallocatechin
GC : Gallocatechin
EGCG : Epigallocatechin gallate
OOBF : Oolonghomobisflavan
Introduction
The Coronavirus Disease 2019 (COVID-19) pandemic, caused by the SARS-CoV-2 virus, has been and continues to be an unprecedented global health challenge. Despite the significant contribution of currently available vaccines in mitigating disease severity and spread, a small percentage of the population exhibits a reduced or even absent immune response. Furthermore, the continuous emergence of new viral variants with mutations in the Spike protein constantly underscores the need to develop alternative or complementary therapeutic approaches capable of acting directly on the virus.
The Spike (S) protein of SARS-CoV-2 is a crucial molecular target. It mediates the binding of the virus to host cell receptors (primarily ACE2) and the subsequent fusion of viral and cellular membranes, representing the initial and fundamental step for viral entry. The Spike glycoprotein is a Class I fusion protein that comprises two main regions, known as S1 and S2, which are responsible for these two distinct functions. The S1 region contains the receptor- binding domain (RBD), which is directly responsible for binding to cellular surface receptors. Its essential role makes it a strategic vulnerability for therapeutic intervention, encompassing both vaccine development and targeted antiviral drug design.
In this context, computational studies, particularly virtual screening techniques such as molecular docking, offer an efficient and cost- effective approach for novel drug discovery. These methods enable the rapid exploration of virtual libraries containing thousands of compounds, identifying those with the highest binding potential to a specific protein target. This significantly reduces the time and resources required compared to traditional experimental approaches. The present study aims to identify novel potential ligands for the SARS-CoV-2 Spike protein, focusing on both natural compounds derived from plant matrices like roasted cocoa, Hypericum perforatum (Hypericum), Hedera helix (ivy), and Salvia officinalis (sage), as well as approved or experimental antiviral drugs. To achieve this, an exhaustive computational screening was conducted using molecular docking (both conventional and covalent approaches) on the trimeric structure of the Spike protein (e.g., PDB ID: 6VSB, in its prefusion conformation). The objective was to identify molecules capable of effectively interacting with the binding site or with key residues, such as cysteine 136. This approach seeks to provide promising candidates for future experimental investigations, thereby contributing to the ongoing search for new effective therapeutic strategies against COVID-19.
Materials and Methods
This computational study was conducted using the Flare software suite (Cresset, Cambridge, UK), an advanced platform for molecular modeling and virtual screening. Specifically, the Docking module, supporting both conventional and covalent approaches, was employed for molecular anchoring.
Receptor and Ligand Preparation
The three-dimensional structures of the SARS-CoV-2 Spike protein trimer were obtained from the Protein Data Bank (PDB) and used as models. Two distinct conformations were utilized:
- PDB ID: 6VSB (Prefusion 2019-nCoV spike glycoprotein with a single receptor-binding domain up), representing the pre-fusion conformation of the full trimeric Spike protein.
- PDB ID: 6LZG (Structure of novel coronavirus spike receptor-binding domain complexed with its receptor ACE2), representing the receptor-binding domain (RBD) of the Spike protein bound to its host receptor, Angiotensin-converting enzyme 2 (ACE2).
Before their use in docking simulations, the structures of both proteins were prepared. This involved removing non-essential water molecules, optimizing the positions of hydrogen atoms, and assigning correct protonation states to amino acid residues, considering physiological pH. For the 6LZG complex, the binding interface between the Spike RBD and ACE2 was considered as the docking site. These two distinct conformations of the Spike protein (or its RBD) expose different residues at their binding interfaces, thus potentially accommodating ligands with varying affinities and interaction profiles.
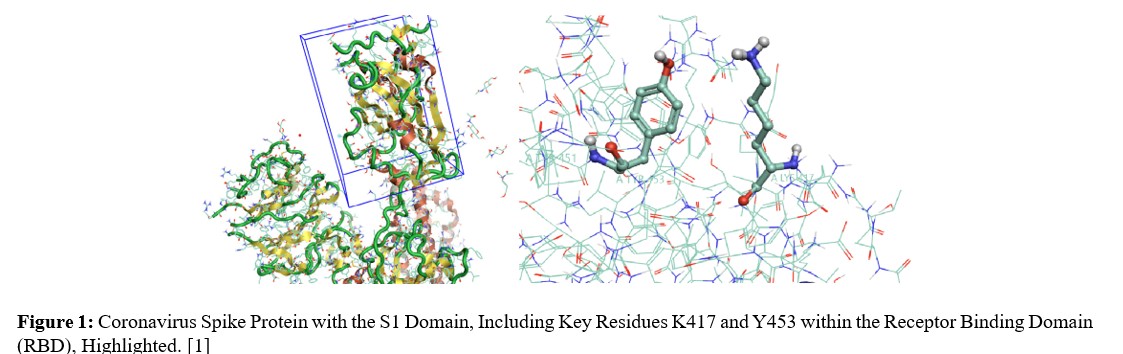
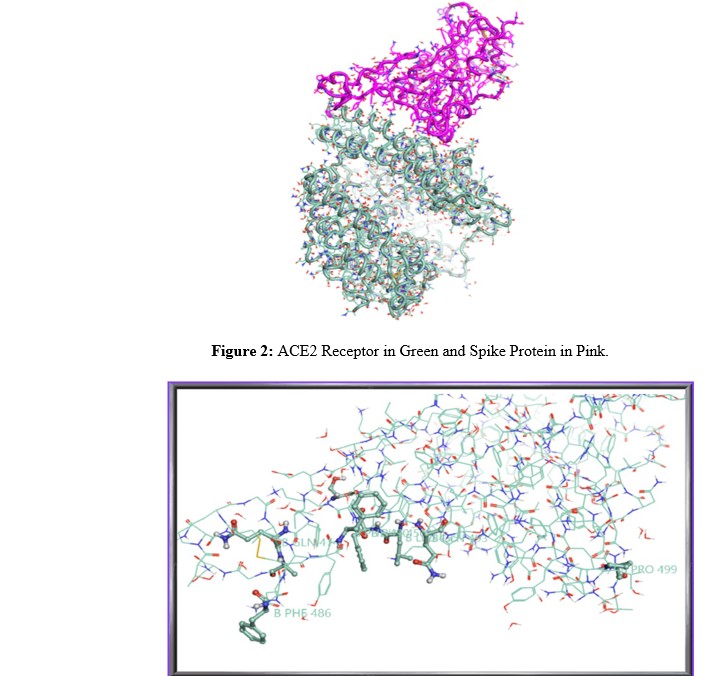
Figure 3: Detailed View of the Spike Protein and Amino Acid Residues Involved in Binding with Angiotensin-Converting Enzyme 2 (ACE2).
Ligands were selected from two main categories:
- Natural Compounds: A library of natural compounds was constructed, comprising extracts and active principles identified from plant matrices of interest, specifically roasted cocoa (Theobroma cacao) [Table 1], Hypericum (Hypericum perforatum) [Table 3], ivy (Hedera helix) [Table 2], and sage (Salvia officinalis). Cacao was chosen due to its diverse chemical composition, including catechins, alkaloids, flavonoids, and Hypericum and ivy were included for their documented antiviral properties, with ivy specifically approved in supplements for respiratory tract affections [2,3]. Compound libraries for each species were generated by downloading compounds in SDF format from PubChem, referencing compounds identified in specific studies [4-6]. Compounds from hops (Humulus lupulus) and wormwood (Artemisia absinthium) were also screened, but they did not yield significant binding scores.

- Antiviral and Reference Drugs: A selection of widely studied, discussed, or utilized antiviral drugs (even those with variable clinical outcomes for COVID-19) was included in the screening. These drugs were curated from various public sources and databases to represent a heterogeneous set of molecules with known antiviral activity or relevance to SARS- CoV-2 research. Examples include viral protease inhibitors (e.g., lopinavir, ritonavir), RNA polymerase inhibitors (e.g., remdesivir, favipiravir, ribavirin), and drugs with presumed antiviral or immunomodulatory activity (e.g., chloroquine, hydroxychloroquine). [Include your table of drugs here or refer to it, e.g., "See Table 4 for the complete list of selected drugs."]. The inclusion of these drugs served to establish a benchmark for binding scores and to compare the affinity of natural compounds with molecules whose biological activity and safety profiles are already partially characterized. Although the primary mechanism of action for many of these drugs is not directly targeted at the Spike protein, their computational screening against this target allowed for the exploration of potential unexpected interactions and the evaluation of their affinity relative to the natural compounds.
- Molecular Docking Methodology
Virtual screening was conducted using two complementary molecular docking approaches:
- Conventional (Non-Covalent) Docking (Structure-Based Screening): This methodology was employed to evaluate the non-covalent binding affinity between ligands and the Spike protein. The active site was defined around the binding region of the S1 subunit and its interface with the ACE2 receptor (refer to Figure 1). The docking parameters were set to allow ligand flexibility and limited flexibility for some key residues within the binding The docking score was utilized as the primary metric to assess binding affinity, with more negative scores indicating stronger binding. The types of interactions (e.g., hydrogen bonds, hydrophobic interactions, electrostatic interactions) between the ligand and the protein were also analyzed.
Classification score: Optimized to accurately predict the 3D positions of docked ligands.
dG: Optimized to accurately estimate the protein-ligand binding
energy, assuming the correct orientation of the ligand.
VS: Optimized for maximum efficiency in virtual screening experiments, discriminating between active and inactive compounds.
- Covalent Docking: This approach was employed to identify compounds capable of forming a stable and irreversible (or quasi-irreversible) covalent bond with a specific residue on the Spike protein, a mechanism of action often associated with potent For this study, cysteine 136 was selected as the target for covalent bond formation.
Rationale for Cysteine 136 Selection
The selection of cysteine 136 (Cys136) was based on several considerations:
- Accessibility and Exposure: Cys136 is an exposed residue on the surface of the Spike protein, making it accessible for interaction with external ligands, unlike other cysteines that may be involved in internal disulfide bonds within the protein structure and thus less reactive or available. [Content Suggestion]: The general explanation about cysteine's role in protein folding and enzyme catalysis, while true, is quite broad for a Methods section. Keep this section focused specifically on why Cys136 was chosen for your study. You can remove sentences like "I residui cisteinici sono residui chiave nella funzionalità delle proteine in genere..." to maintain conciseness and focus on the experimental design.
- Thiol Reactivity: The thiol group (-SH) of cysteines is known for its high nucleophilicity and reactivity, particularly towards electrophiles such as nitrile groups present in some natural and synthetic compounds.
- Sulfide-Nitrile Reaction: The conjugation reaction between a thiol group (such as that of cysteine) and a nitrile group is particularly relevant. This reaction proceeds via the initial formation of a thioimidate adduct which, in the presence of an adjacent amine (such as from the protein backbone or a nearby residue), can cyclize into a stable, and in some cases irreversible, thiazoline ring (e.g., through a Michael-type reaction or cycloaddition). This reaction is well-documented in the literature for its selectivity, efficiency, and often its ability to proceed at ambient temperature [7]. This reactivity has also been utilized in analytical recognition assays for the formation of stable covalent bonds.
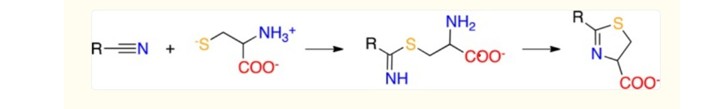
Scheme 1: Reaction between Nitrile-Containing Compounds and Cysteine Via Cycloaddition. Adapted from Berteotti et al., 2014 [7].
Potential Inactivation Mechanism: Covalent binding to a crucial residue of the Spike protein could lead to its stable functional inactivation, thereby preventing virus-host interaction.
The covalent docking process simulated the formation of the bond between reactive ligands (e.g., those containing nitrile groups or other electrophilic moieties) and the thiol group of Cys136, evaluating the binding energy and stereochemical plausibility.
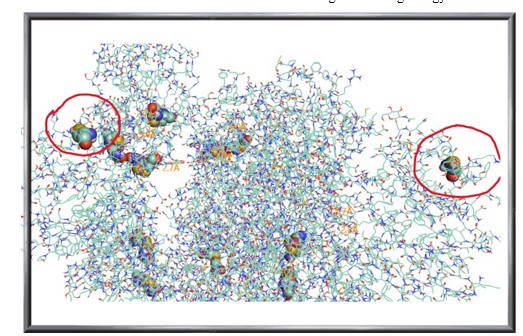
Figure 4: Cysteine Residues of the Spike Protein: Unbound (Circled in Red) and those Involved in Disulfide Bonds (Represented as Spheres)
Results Analysis and Selection Criteria
Compounds were classified based on their docking scores (where more negative scores indicate a higher binding affinity) and the quality of the interactions established with key protein residues. During the molecular docking process, for each ligand tested, multiple possible conformations within the receptor binding pocket were generated and evaluated. The software calculates an affinity score for each generated pose (binding pose). Only the conformation presenting the most favorable binding score (i.e., the lowest, or most negative, depending on the metric used by the software to indicate stronger affinity) was considered the 'optimal' or 'best' pose and used for subsequent results analysis. Visual analyses of the binding poses were performed to confirm the plausibility of the interactions and to identify potential interactions with residues critical for the Spike protein's function. Only compounds with the best scores and significant interactions with the protein were selected for in-depth analysis and discussion. The sum of scores (rank score, delta G, virtual screening score) was considered to evaluate which molecules best fit the protein.
Results
Docking of Natural Compounds on the Spike Protein
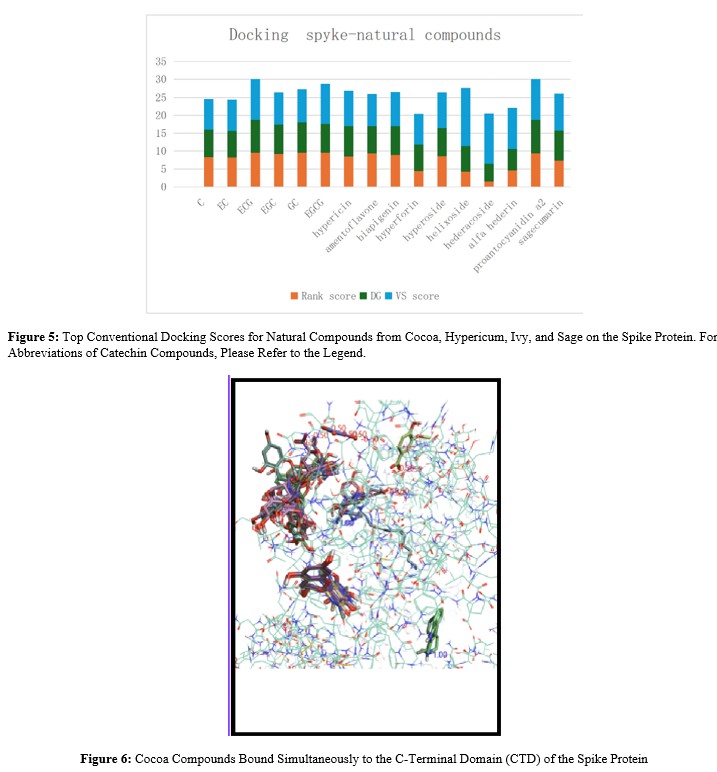
Docking simulations were performed for compounds identified in cocoa (Table 1), ivy (Table 2), and Hypericum (Table 3). The results are illustrated in Figure Epicatechin gallate and procyanidin A2 achieved the highest scores based on the sum of three partial scores (rank score, delta G, and VS score). Compounds from wormwood (Artemisia absinthium) and hops (Humulus lupulus) were also screened but did not yield significant binding scores.
As observed in Figure 7, two compounds from ivy, hederin and hederacoside, being voluminous compounds, appear to cover the entire involved surface, indicating favorable p harmacodynamics. However, due to their large molecular weights (472 Da and 1205 Da, respectively), they are predicted to have unfavorable pharmacokinetics (absorption, distribution, metabolism, excretion, i.e., ADME profile), despite some antiviral drugs also being large molecules (e.g., ritonavir, 720 Da). Some compounds from Hypericum (Figure 8) exhibited gooddocking scores and showed a favorable ADME profile (data not shown). Sagecoumarin from sage also presented a promising score. Docking simulations performed on the pre-fusion Spike protein and the ACE2-bound Spike protein yielded substantially similar binding score values.

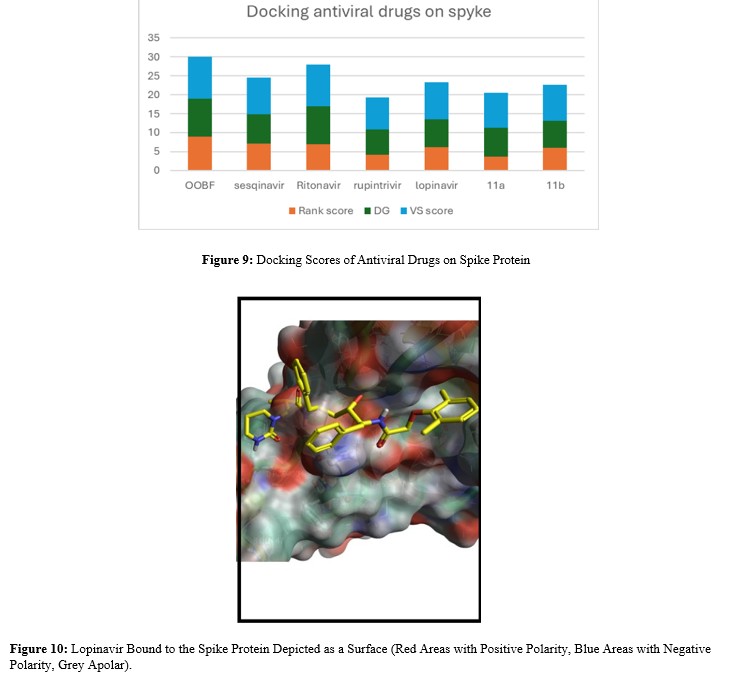
- Docking of Antiviral Compounds on Spike Protein Antiviral compounds from various sources and with diverse mechanisms were tested, given that the Spike protein does not have small-molecule drugs specifically known to target it.
Oolonghomobisflavan (a dimer of epigallocatechin), which exhibits antiviral activity, achieved the best docking scores among these compounds [8]. It is a highly polar molecule with a Topological Polar Surface Area (TPSA) of 395 Ų. For orally bioavailable drugs, a common reference value for TPSA is around 60 Ų, suggesting that Oolonghomobisflavan is unlikely to readily cross biological membranes and therefore has an unfavorable pharmacokinetic profile for systemic absorption. Ritonavir was the antiviral drug that yielded the best docking scores. While Ritonavir's primary role in COVID-19 treatment (e.g., in combination with Nirmatrelvir) is as a pharmacokinetic booster that reduces hospitalizations and prevents long-term COVID-19, its high docking score in this study suggests a potential interaction with the Spike protein [9].
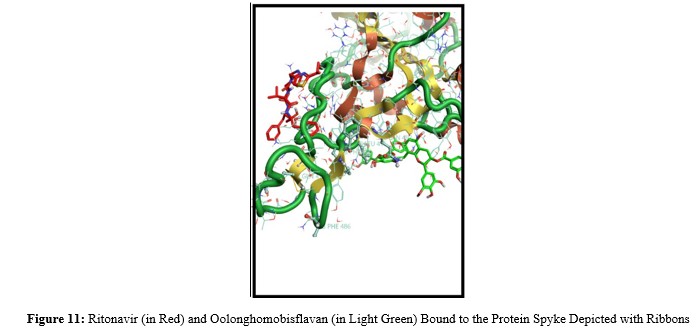
Covalent Docking on Spike Protein
Covalent docking simulations were performed on the Spike protein, targeting not the entire molecule, but specifically cysteine 136, as it is the only cysteine residue not involved in a disulfide bond and thus most suitable for this type of interaction. Natural compounds containing nitrile groups (Table 5) and approved or experimental drugs containing nitrile (Table 6) were tested [10,11]. The best results were obtained for Epanolol, a beta-blocker, and
Vilazodone, an antidepressant, which achieved the highest scores among the drug set (Figure 14). For the natural compounds (Figure 13), Bauhinin and Bursatellin yielded the best scores. Bauhinin is a natural phytochemical found in the Bauhinia genus, characterized by a flavonoid glycoside structure. The bursatellin-oxazinine family is a series of nitrile-containing marine natural products derived from tyrosine, originating from gastropod and bivalve mollusks.

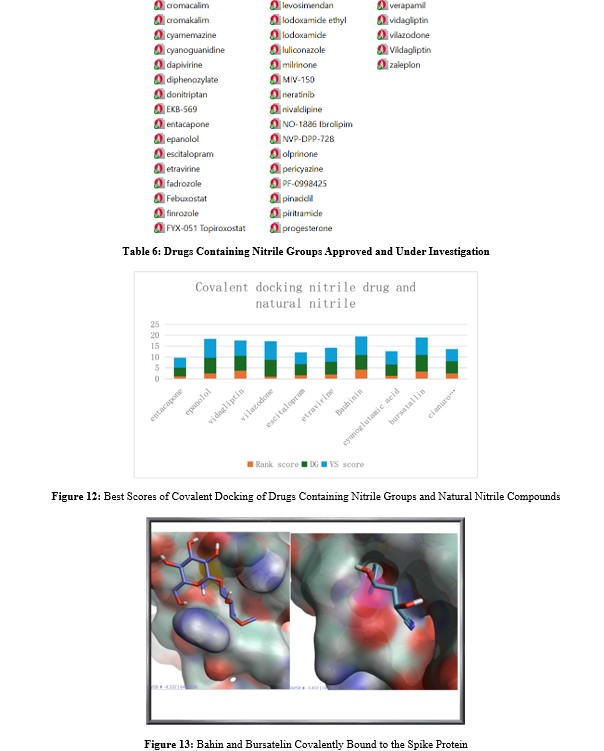
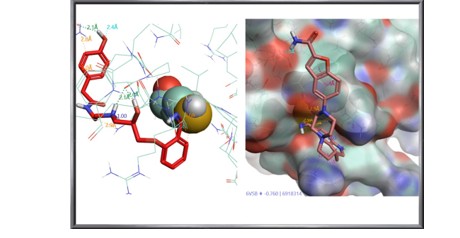
Figure 14: Epanolol and Vilazodone, Covalently Linked to the Spyke Protein (Cysteine 136 in the Left Image is Depicted with Spheres)
Discussion
In classical pharmacology, the activity of a single compound on a protein (receptor or otherwise) is typically evaluated. However, when considering the intake of a natural species such as cocoa, ivy, or Hypericum, multiple compounds are simultaneously present. These compounds can bind to different positions on the target protein, depending on their individual affinities. In the context of antiviral compounds, whether natural or synthetic, the goal is to bind to the viral target and, in this case, inhibit the Spike protein. It would be particularly interesting to experimentally test whether mixtures of natural compounds, such as hypericin, amentoflavone, and biapigenin—which demonstrated the best scores for Hypericum perforatum—could inhibit the virus.
Upon observation of Figures 7 and 8, it is evident that amino acids within the Spike protein that are crucial for binding with the Angiotensin-converting enzyme 2 (ACE2) receptor are occupied by interactions with the natural compounds. This suggests a potential mechanism for interfering with viral entry. Furthermore, existing studies on the use of antiviral drug mixtures have shown more satisfactory results compared to the use of a single molecule, supporting the potential of multi-component approaches [12]. A relevant study on antidepressant use in COVID-19 indicated significant associations between citalopram, escitalopram, venlafaxine, desvenlafaxine, mirtazapine, doxepin, and vilazodone, and a reduced risk of worse COVID-19 outcomes [13]. This clinical observation aligns with our computational findings regarding the docking potential of Vilazodone on the Spike protein.
Conclusion
This study performed molecular docking simulations of key compounds found in several plant matrices (roasted cocoa beans, Hypericum, ivy, wormwood, sage), alongside currently used and experimental antiviral drugs. Additionally, a specific analysis was conducted on drugs and natural products containing nitrile groups, which are capable of forming covalent bonds with cysteine residues on proteins.
The results from conventional docking revealed good affinity for the Spike protein with compounds such as:
- Catechins (catechin, epicatechin, epigallocatechin gallate, epicatechin gallate, epigallocatechin, gallocatechin) present in cocoa (Theobroma cacao).
- Procyanidin A2, amentoflavone, biapigenin, hyperoside, and hypericin from St. John's Wort (Hypericum perforatum).
- Ivy (Hedera helix) compounds including hederacoside, alpha- hederin, hederagenin, elixoside, and procyanidin A1. These compounds yielded high scores and demonstrated binding over a large surface area, interacting with multiple amino acids involved in the Spike protein's binding to the ACE2
For antiviral drugs, both currently in use and experimental, higher activity was observed when docked to the receptor- bound conformation of the Spike protein, with Ritonavir notably showing strong scores. Oolonghomobisflavan-A and -B also exhibited excellent scores and are reported to inhibit COVID proteases [14]. Regarding covalent docking with nitrile-containing compounds forming bonds with cysteine groups, while these compounds successfully bound covalently to Cysteine 136 (the only exposed cysteine not involved in a disulfide bond within the three homotrimers of the Spike protein), the overall scores were not exceptionally high compared to the best conventional binders. Specifically, Bauhinin and Bursatellin were the top-scoring natural compounds, and Epanolol and Vilazodone were the top-scoring drugs in this covalent approach. Based on these findings, it would be interesting to investigate clinically whether patients undergoing treatment with Vilazodone (an antidepressant drug) exhibited a lower incidence of COVID-19 compared to the general population [15-19].
Protein Visualizations
Protein representations with bound compounds were created using Flare.
Acknowledgements
The authors would like to thank Cresset for providing academic licenses for Flare.
Funding
This project was self-funded.
Statement on the Use of Artificial Intelligence
Gemini (version 2.5 Flash) was utilized for structuring the manuscript from draft, as well as for translation and grammatical correction.
Conflict of Interest Statement
The authors declare no conflict of interest.
Graphs
Graphs were generated using Microsoft Excel (academic license).
References
- Yi, , Sun, X., Ye, J., Ding, L., Liu, M., Yang, Z., ... & Sun, B. (2020). Key residues of the receptor binding motif in the spike protein of SARS-CoV-2 that interact with ACE2 and neutralizing antibodies. Cellular & molecular immunology, 17(6), 621-630.
- Jacobson, M., Feinman, L., Liebes, L., Ostrow, N., Koslowski, V., Tobia, A., ... & Prince, A. M. (2001). Pharmacokinetics, safety, and antiviral effects of hypericin, a derivative of St. John's wort plant, in patients with chronic hepatitis C virus infection. Antimicrobial agents and chemotherapy, 45(2), 517-524.
- Song, , Yeo, S. G., Hong, E. H., Lee, B. R., Kim, J. W., Kim, J., ... & Ko, H. J. (2014). Antiviral activity of hederasaponin B from Hedera helix against enterovirus 71 subgenotypes C3 and C4a. Biomolecules & Therapeutics, 22(1), 41.
- Cabras, , Martelli, A. (2004). Chimica degli Alimenti. Padova: Piccin-Nuova Libraria, ISBN: 978-88-299-1696-2.
- Yasmeen, S., Usman, R., Ayaz, S., Qamar, F., Zainab, S., Sadia, H., ... & Faridi, Z. U. (2020). Quality and Potency Analysis of IVY Leaf Extract. RADS Journal of Pharmacy and Pharmaceutical Sciences, 8(1), 58-62.
- Butterweck, , Schmidt, M. (2007). St. John's wort: Role of active compounds for its mechanism of action and efficacy. Wien Med Wochenschr,157(13-14), 356-61.
- Berteotti, , Vacondio, F., Lodola, A., Bassi, M., Silva, C., Mor, M., & Cavalli, A. (2014). Predicting the reactivity of nitrile-carrying compounds with cysteine: a combined computational and experimental study. ACS medicinal chemistry letters, 5(5), 501-505.
- Chojnacka, K., Skrzypczak, D., Izydorczyk, G., Mikula, K., Szopa, D., & Witek-Krowiak, (2021). Antiviral properties of polyphenols from plants. Foods, 10(10), 2277.
- Saheb Sharif-Askari, F., Ali Hussain Alsayed, H., Saheb Sharif-Askari, , Al Sayed Hussain, A., Al-Muhsen, S., & Halwani, R. (2024). Nirmatrelvir plus ritonavir reduces COVID-19 hospitalization and prevents long COVID in adult outpatients. Scientific Reports, 14(1), 25901.
- Fleming, (1999). Nitrile-containing natural products. Natural Product Reports, 16(5), 597-606.
- Fleming, F., Yao, L., Ravikumar, P. C., Funk, L., & Shook,
- C. (2010). Nitrile-containing pharmaceuticals: efficacious roles of the nitrile pharmacophore. Journal of medicinal chemistry, 53(22), 7902-7917.
- Merry, C., Barry, M. G., Mulcahy, F., Ryan, M., Heavey, J., Tjia, F., ... & Back, D. J. (1997). Saquinavir pharmacokinetics alone and in combination with ritonavir in HIV-infected patients. Aids, 11(4), F29-F33.
- Rahman, M., Mahi, A. M., Melamed, R., & Alam, M. A (2023). Effects of antidepressants on COVID-19 outcomes: retrospective study on large-scale electronic health record data. Interactive Journal of Medical Research, 12(1), e39455.
- Ullah, , Ullah, S., Halim, S. A., Waqas, M., Ali, B., Ataya,
- S., ... & Al-Harrasi, A. (2024). Identification of new pharmacophore against SARS-CoV-2 spike protein by multi- fold computational and biochemical techniques. Scientific Reports, 14(1), 3590.
- Zhu, , He, G., Yin, Q., Zeng, L., Ye, X., Shi, Y., & Xu, (2021). Molecular biology of the SARsâ?ÂÃÃÃÃÂ????ÂÃÃÃÂ???ÂÃÃÂ??ÂÃÂ?ÂÂCoVâ?ÂÃÃÃÃÂ????ÂÃÃÃÂ???ÂÃÃÂ??ÂÃÂ?ÂÂ2 spike protein: A review of current knowledge. Journal of medical virology, 93(10), 5729-5741.
- Wang, , Zhang, Y., Wu, L., Niu, S., Song, C., Zhang, Z.,.. & Qi, J. (2020). Structural and functional basis of SARS- CoV-2 entry by using human ACE2. Cell, 181(4), 894-904.
- Bhardwaj, K., Singh, R., Sharma, J., Rajendran, V., Purohit, R., & Kumar, S. (2021). Identification of bioactive molecules from tea plant as SARS-CoV-2 main protease inhibitors. Journal of Biomolecular Structure and Dynamics, 39(10), 3449-3458.
- Paul, S., Islam, R., Parves, M. R., Mamun, A. A., Shahriar, I., Hossain, M. I., ... & Halim, M. A. (2022). Cysteine focused covalent inhibitors against the main protease of SARS-CoV-2. Journal of Biomolecular Structure and Dynamics, 40(4), 1639-1658.
- Holendova, B., & Plecita-Hlavata, L. (2023). Cysteine residues in signal transduction and its relevance in pancreatic beta cells. Frontiers in endocrinology, 14, 1221520.