
Journal of Clinical & Experimental Immunology(JCEI)
ISSN: 2475-6296 | DOI: 10.33140/JCEI
Impact Factor: 1.9

Research Article - (2026) Volume 11, Issue 1
Benzoate Drugs for Traumatic Brain Injury
2Department of Neurological Sciences, Rush University Medical Center, Chicago, USA
Received Date: Feb 19, 2026 / Accepted Date: Mar 06, 2026 / Published Date: Mar 14, 2026
Copyright: ©2026 Kalipada Pahan, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Citation: Pahan, K., Pahan, S. (2026). Benzoate Drugs for Traumatic Brain Injury. J Clin Exp Immunol, 11(1), 01-10.
Abstract
Traumatic brain injury (TBI) is an acute disorder with prolonged complexities that affirm to be the leading cause of deaths and disabilities in US. The pathophysiology of TBI comprises of multiple complex processes. An impactful insult results in tissue damage, neuroinflammation, impaired cerebral blood flow, BBB disruption, and demyelination, leading to neurodegeneration and functional impairments. There are no specific treatments for TBI. Recent studies highlight the potential efficacy of the following benzoates for attenuating TBI pathologies in rodents:
• Sodium benzoate, a metabolite of cinnamon, a widely used food additive and a Food and Drug Administration (FDA)-approved drug for glycine encephalopathy
• Glyceryl tribenzoate, FDA-approved flavoring element being used in food and food packing productions
In this review, we have made a genuine attempt to evaluate the significance of these two benzoates as prospective therapeutic options for TBI
Keywords
Traumatic Brain Injury, Sodium Benzoate, Glyceryl Tribenzoate, Glial Activation, Neurodegeneration, Oxidative Stress, Cognitive Dysfunction, Neuroprotection, Drug Repurposing
Introduction
Traumatic brain injury (TBI), also known as an intracranial injury, is caused by sudden jolt, bump or blow on the head due to fall, automobile crash, firearms, sports related injuries, and exposure to explosive at war [1,2]. Studies have shown that nearly half of the total TBI-related hospitalization is associated to fall and that firearm-related suicide is the most common cause of TBI-initiated death in US [3]. CDC report on TBI indicates that there were over 2.1 million emergency department visits for injuries from motor vehicle crashes and that about 41,000 people died in motor vehicle crashes in US in.2020. It has been reported that 5.3 million Americans are disabled of which 80,000 victims suffer from long term disability and about 70,000 deaths reported annually. TBI has significantly impacted the wellbeing of millions globally in all age groups, leading to temporary, permanent disabilities and untimely deaths [4]. To recoup with the devastating outcome of TBI, about $76.5 billion is attributed for care and loss of productivity every year. Studies show that the pathophysiology of TBI include neuroinflammation followed by neurodegeneration resulting in locomotive, cognitive dysfunctions, and neuronal deaths [5,6]. Regrettably, treatments available for TBI can only treat the symptoms to a certain extent and thereby only rest and medications like antianxiety, anticoagulant, anti-convulsant, antidepressants and muscle relaxants are prescribed for stabilization till surgery, which is evidently a perilous procedure. Therefore, there is an absolute need to investigate for a safe, nontoxic, conveniently administrable, and economic therapeutic agent to potently curb the devastating outcomes of TBI. In rodents, TBI like pathologies are induced by weight drop technique, controlled cortical impact (CCI), blast injury, and fluid percussion injury [7-10]. Studies show NaB, an FDA-approved treatment for urea cycle disorder and glycine encephalopathy in children, and GTB, a flavoring agent approved by Flavor and Extract Manufacturers Association (FEMA), can competently inhibit neuroinflammation and prevent neurodegeneration by improving locomotor and cognitive dysfunctions in CCI-induced mouse model of TBI [11,12]. Our investigation focuses on the neuroprotective efficacy of these two benzoates and evaluates the potential therapeutic possibilities for TBI treatment.
Clinical symptoms of TBI
TBI is an aggressive and amalgamated disease process that leads to impairment of different body functions or even death in all age group of the population, especially in older adults with worse functional outcomes and higher mortality [1,2]. TBI can be classified by the nature of the injury, penetrating (open) and non-penetrating (closed) TBI. Penetrating TBI is caused by sharp weapon or bullet entering the skull and affecting the part of the brain that encounters the insult. Non- penetrating (closed) TBI usually is caused by an external insult on the head acting on the whole brain exhibiting devastating clinical symptoms [13]. TBI can also be classified depending on the severity of the impact of the insult ranging from mild, moderate to severe. Clinical symptoms of mild TBI are marked by brief loss of consciousness, headache, confusion, dizziness, blurred vision and ear ringing, bad taste in mouth, fatigue, mood changes, sleep pattern trouble, and memory issues [7]. Moderate to severe TBI symptoms are characterized by headache getting worse, repeated vomiting and nausea, convulsion, seizures, numbness, weakness of arms and legs, loss of coordination, increase in confusion, restlessness and agitation, and not being able to wake up from sleep or coma and death.
Cognitive Deficit
It is caused by impairments of the network of neurons of the temporal and frontal lobes of the brain that primarily affect learning, memory, perception, and problem-solving abilities. Cognitive dysfunction is one of the problematic outcomes among TBI survivors [14]. Similar clinical symptoms of cognitive deficits are observed in one of the most prevalent neurodegenerative disease, Alzheimer’s disease (AD) [15]. Clinical studies on postmortem brains of TBI victims have shown an increased level of hyperphosphorylated tau (P-tau) and amyloid beta and TDP 43 deposits, indicating AD-like pathologies in TBI condition [16]. Studies also indicate that older adults with a history of moderate TBI are at 2.5 times greater risk of developing AD than seniors with no history of TBI and that persons with a history of severe TBI have 4.5 times greater AD risk than non-TBI population [16].
Psychological Problems
Mood disturbance, depression and anxiety are neuropsychiatric disorders regulated by frontal cortex, basal ganglia and temporal lobes of the brain that significantly impact the behavioral and emotional aspect and affect recovery, functional ability, and daily life activities of the concern individual [17]. TBI survivors experience neuropsychiatric dysfunctions at a much higher rate than general population. Depression is a common mental disorder characterized by feeling of sadness and hopelessness, which persist and eventually interfere with daily normal activities of the affected individual. TBI and depression have a close association. Studies show about 6-77% of TBI patients experience depressive disorder, among which 25-50% diagnosed with first-year post-TBI depressive disorder and 26-64% have life-time risk of depression development [18]. Similarly, anxiety is also a TBI consequence that affects about 20% of TBI patients within one year of injury [19]. It has been shown that TBI individuals are 1.9 times more likely to develop anxiety symptoms as compared to normal individuals [19].
Post-Traumatic Stress Disorder (PTSD)
PTSD is a mental disorder caused by experiencing or witnessing terrifying incidents. It is characterized by dysphoric mood, anxiety, sleep disturbances, irritability, anger, poor concentration, fatigue, and upsetting memory of the terrible event. Epidemiological data confirms that PTSD development is significantly prominent among mild TBI survivors [20]. Moreover, rate of PTSD following a brain injury is higher in military service men than in civilians due to their prolonged exposure to combat. About 35% returning military with a mild brain injury experience PTSD [21]. Proper diagnosis and treatment are needed for managing these conditions.
Locomotor Impairment
Across the spectrum of TBI from mild to moderate to severe, patients suffer from locomotor impairment. In some TBI patients, shearing of axons and excess neuroinflammation initiate the loss of communication of signals hindering coordination and resulting in tremor, akinesia, and postural instability, triad symptoms that are hallmarks of Parkinson’s disease (PD), the most common neurodegenerative movement disorder caused by the demise of dopaminergic neurons present in the substantia nigra compacta region of the midbrain. It has been shown that TBI is associated with a risk for Lewy body accumulation and parkinsonism [22,23]. LRRK2 is connected with late onset of PD and recent studies indicate the activation of LRRK2 in TBI mice [24]. Studies also demonstrate the upregulation of amyloid precursor protein (APP), hyperphosphorylation Tau and TAR DNA-binding protein 43 (TDP-43) in TBI patients, which are closely linked to PD. It has been also shown that TBI leads to PD-like pathologies in mice [23,25]. According to Delic et al., veterans with a history of mild TBI are at 56% higher risk for developing PD later in life and the PD risk grows with increase in TBI severity. Therefore, TBI is definitely a risk factor for PD [23].
Pathological hallmarks of TBI
Progression of TBI can be distinguished in 3 phases -acute, post- acute and chronic. While the acute phase initiates immediately upon insult to a week, the post-acute phase lasts from 1 week to 1 month. On the other hand, the chronic phase lasts from 1 month to years. Here, we will describe key pathological features that are present in different phases of TBI.
Neuroinflammation
Immediately after an insult, the brain’s immune system gets stimulated and initiates neuroinflammation to combat and heal. Therefore, neuroinflammation has a beneficial window and is not bad all the time. However, this essential process sometimes prolongs, leading to uncontrolled with upregulated production of proinflammatory molecules like TNFα, IL-6, IL- 1β, nitric oxide (NO), etc. [26-32]. Then the chronic unrestrained neuroinflammation becomes detrimental for the brain. It has been reported that TNFα is capable of promoting the actin stress fiber formation and downregulating the tight junction protein occludin, resulting in retraction of endothelial cells and creation of gaps in the BBB [33]. This allows the compromised BBB to facilitate the infiltration of different cytotoxins into the brain causing neurodegenerative damage.
Demyelination
Although previous studies on TBI stressed on pathological modifications in neuronal cells within the gray matter, recent studies have highlighted the equal importance of white matter integrity in long-term recovery from TBI-related damages [34]. Demyelination is an important feature of white matter injury that is characterized by the loss of the myelin sheath [35,36]. Oligodendrocytes are the primary cells responsible for the generation and maintenance of myelin sheath under normal conditions and for remyelination after axonal damage [37-39]. However, the loss of oligodendrocytes is known to be a significant factor underlying demyelination after CNS injury as myelinating oligodendrocytes are highly vulnerable to ischemic or traumatic insults [40-42]. Accordingly, using a large-scale cohort study design, Kang and Lin have examined the risk for multiple sclerosis (MS) following a TBI and found increased risk of MS after TBI [43].
Axonal Injury
In white matter tracts, axons are encapsulated by oligodendrocyte- generated myelin. The axon- derived signals are required for the proliferation, migration, survival, and differentiation of oligodendrocytes [44,45]. In addition, oligodendrocytes are also actively involved in detecting axonal energy needs and preserving axonal integrity [46]. Therefore, once an axon is demyelinated, its ability to transmit action potentials and its energy supply are severely impaired, causing the exposed nerve fiber to be highly susceptible to degeneration during TBI. This hinderance of communication results in loss of physical and mental coordination or disabilities among TBI patients. Although the etiology of MS is unknown, in many MS patients, demyelination of axons is seen [47]. A systematic review and meta-analysis have delineated that the risk of MS increases among people with a history of head trauma [48].
Intracerebral Hematoma and Edema
Intracerebral hematomas and edema are the most damaging outcome of TBI [49]. Intracerebral hematoma is the accumulation of blood within the brain due to coalescence of contusion upon TBI. This results in increased intracranial pressure leading to brain damage, unconsciousness or even death. Edema is the swelling caused by fluid leak in blood vessel into nearby tissues due to the rapture of the vessels during the brain injury. Hematoma and edema result in brain compression and increase in ICP and BBB disruption, subsequently developing enlarge lesion cavities in the temporal lobes. This is probably the leading cause to mortality and mobility for TBI patients.
Oxidative Stress
Oxidative stress is caused by an imbalance between the reactive oxygen species (ROS) and the antioxidant defense system in the cells. The overproduction of ROS\ indulges in harmful chains of oxidation reactions resulting in the breakdown of cells and leading to serious neurological diseases like MS, AD, PD, HD, etc. [50- 53]. Studies have shown the involvement of oxidative stress in the pathogenesis of TBI [54]. The level of CSF malondialdehyde (MDA), oxidative stress marker, is elevated within 2-3 hours of TBI and remain elevated for several days post injury [55]. Moreover, the activity of antioxidant defense enzyme superoxide dismutase (SOD) decreases 24 hours post TBI and remain so 7 days post severe TBI [54]. Moreover, oxidative stress is known to trigger the production of proinflammatory molecules via activation of NF-κB [56]. However, studies have shown that NaB treatment can reduce LPS-induced production of ROS in mouse microglia [57]. NaB also inhibits ROS production from microglia stimulated by fibrillar amyloid-β (an etiological reagent for AD) and 1 methyl-4- phenylpyridinium ion (a Parkinsonian toxin) [57]. Small G protein p21rac is an important member of NADPH oxidase, a five-subunit enzyme complex that catalyzes the production of superoxide radical [58]. NaB inhibits the activation of p21rac via reducing the geranylation pathway and thereby decreases the production of ROS from activated microglia [57]. Oral administration of NaB also exhibits antioxidant effect in vivo in the brain of 5XFAD mouse model of AD [57]. Following the same mechanism, NaB treatment may also reduce oxidative stress in the hippocampus under TBI condition.
Sodium benzoate in TBI
NaB is a metabolite of cinnamon, a household spice and flavoring agent [59,60]. NaB is also a food-additive and an FDA-approved drug against urea cycle disorders and glycine encephalopathy[61,62]. CCI-induced mouse model is a reliable model to showcase the symptoms of TBI [10]. Recent study has highlighted the beneficial effect of NaB in CCI-induced TBI mice [12].
Inhibition of Glial Activation
Upon brain injury, glial cells like astroglia and microglia are activated to express higher levels of GFAP and Iba-1, respectively. Moreover, activated glial cells produce a wide variety of proinflammatory molecules like TNFα, IL-1β, IL- 6, IL-8, macrophage inflammatory protein-1 alpha (MIP-1α), NO, etc., which could ultimately lead to neuroinflammation and neurodegeneration [31,63-66]. However, oral NaB treatment leads to decrease in both GFAP positive astrocytes and Iba-1 positive microglia in the cortex and hippocampus of CCI-induced TBI mice [12]. Activated glial cells also express elevated levels of iNOS for the induced production of NO, which sometimes leads to nitrosative stress [31,65,67]. It is encouraging to see that NaB treatment results in the suppression of iNOS in astroglia and microglia in vivo in the brain of TBI mice [12]. It has been described that NaB inhibits the expression of proinflammatory molecules in activated glial cells via suppression of farnesylation of p21ras and activation of NF- κB pathway (Figure 1) [68].
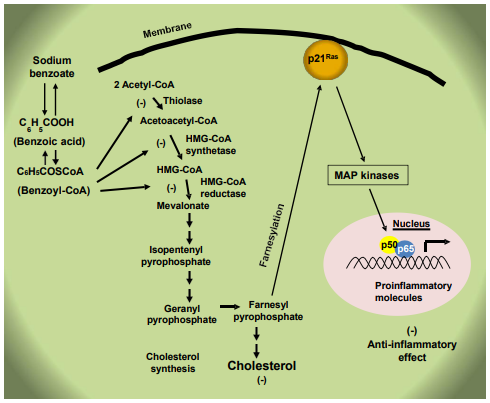
Figure 1: Anti-inflammatory effect of NaB. Cholesterol is synthesized within the cells from acetyl-CoA via multiple steps. NaB forms benzoyl-CoA, which competitively inhibits the first three steps of the cholesterol biosynthesis pathway catalyzed by thiolase, HMG- CoA synthetase and HMG-CoA reductase, respectively to lower the level of farnesyl pyrophosphate. The small G protein p21ras gets attached to the membrane and becomes activated upon farnesylation by farnesyl pyrophosphate. Therefore, by lowering the level of farnesyl pyrophosphate, NaB inhibits the activation of p21ras and associated mitogen-activated protein (MAP) kinase pathway to lower the activation of classical NF-κB p65:p50 heterodimer and the expression of proinflammatory molecules, leading to anti-inflammation.
Reduction of Lesion Volume
TBI often causes enlarged lesion cavity due to the impact of the external insult, which can culminate into serious immunological and neurological disorders with fatal consequences. Considering segmentation of the brain, diffusion, and the damage regions, the lesion region in the brain tissue is usually estimated. The area of the damaged portion is estimated across each slice of MRI after the segmentation followed by estimation of the combined volume of damage through 3D reconstruction. Accordingly, in CCI-induced TBI mice, a typical lesion with enlarged cavity originating from the cortex through the hippocampus and connecting to the lateral ventricle is seen.
Consistent to the suppression of glial activation, oral NaB is capable of reducing the lesion size in the hippocampal region of the brain in TBI mice as detected by the Cavalier stereological techniques [12].
Protection of Memory and Learning
Learning and memory is regulated by the hippocampal region of the brain and the dysfunction of memory and learning is likely to be a major drawback for TBI survivors for the rest of their lives [69]. It has been found that NaB is capable of upregulating plasticity-related molecules, stimulating NMDA- and AMPA- sensitive calcium influx and increasing the spine density of cultured hippocampal neurons [70]. As a result, NaB has been reported to convert poor learning mice to good learning ones [70]. While Barnes maze and T-maze are used for monitoring spatial learning and memory in mice, novel object recognition (NOR) test is used to evaluate short-term memory [71,72]. AD is the most common memory disorder and it has been found that daily oral NaB feeding for 1 month improves memory and learning in 5XFAD mouse model of AD [57]. Similar to 5XFAD mice, the CCI-induced TBI mice also exhibit deficient spatial learning and memory and short-term memory as compared to sham mice [12]. Daily NaB treatment recovers both spatial learning and memory and short-term memory in CCI-induced TBI mice, indicating that NaB is capable of improving hippocampal functions in TBI condition [12].
Reduction of Amyloid Plaques
AD is the most common neurodegenerative disorder causing dementia among the older population around the world [73,74]. Pathologically, AD is characterized by neurofibrillary tangles and neuritic plaques comprising of aggregated Tau and Aβ, respectively [75-78]. It has been shown that oral NaB treatment decreases amyloid plaques from the hippocampus of 5XFAD mouse model of AD [57]. Although amyloid plaques are typically associated to AD and aging, it has been reported that TBI can lead to the formation of amyloid plaques, indicating a possible reason for increased risk of developing AD among TBI individuals [79]. Therefore, by reducing the formation of plaques, NaB may be also beneficial for TBI patients.
Restoration of Dopamine Neurons and Dopamine
Dopamine (DA), a neurotransmitter crucial for various brain functions, is produced by dopaminergic neurons from tyrosine via tyrosine hydroxylase (TH) [80]. Although PD, the most common neurodegenerative movement disorder, is caused by loss of DA, studies have shown that TBI can also cause DA loss leading to neuropsychiatric symptoms and cognitive impairments [81]. However, it has been shown that NaB can increase the mRNA and protein expression of TH and stimulates the production of DA from dopaminergic neuronal cells [82]. Mechanistically, NaB stimulates the transcription of TH in dopaminergic neurons via the activation cAMP response element-binding [82]. Oral feeding of NaB also increases the expression of TH in the nigra, upregulates striatal DA, and improves locomotor activities in normal C57/BL6 and aged A53T-α- syn transgenic mice [82]. Therefore, NaB may also increase the level of DA in the brain of TBI patients for improving movement, reward, cognition, and mood.
Protection of Locomotor Activities
Following TBI, locomotor problems, particularly gait and balance disturbances, are common. This locomotive dysfunction in TBI patients is manifested by weakness as well as loss of neurons ( ). External insults can cause neuronal structural damage, resulting in neuronal loss and locomotor dysfunction in TBI. However, as compared to untreated TBI mice, NaB treatment leads to significant improvement in locomotor activities such as distance traveled, velocity, center frequency, rearing behavior, rotarod performance, gait behavior, and grid performance in TBI mice [12]. In addition to TBI, oral NaB treatment is capable of improving locomotor activities in mouse models of PD, MS and Lewy body dementia [83-85]. Therefore, NaB may exhibit beneficial effects for TBI via suppression of pro-TBI as well as induction of anti-TBI pathways (Figure 2).
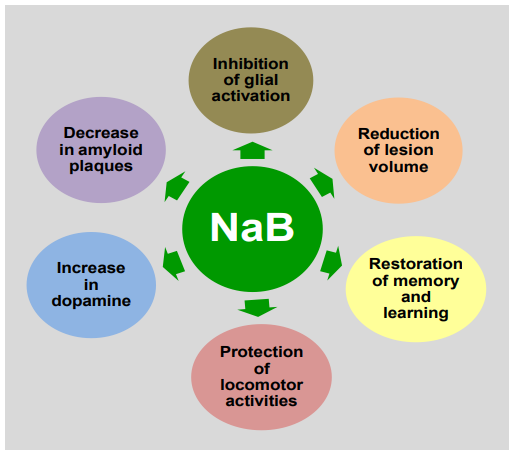
Figure 2: Anti-TBI functions of NaB. In one hand, NaB inhibits glial inflammation, reduces amyloid plaques and decreases lesion volume, and on the other, increases the production of dopamine, upregulates memory and learning and improves locomotor activities to exhibit neuroprotective effects
Glyceryl tribenzoate (GTB) in TBI
GTB is a flavoring reagent used in food and packaging industry [86]. Here, we will discuss neuroprotective effects of GTB in TBI mice.
Attenuation of Neuroinflammation
Since neuroinflammation is an important pathological feature of TBI, a prospective drug should attenuate neuroinflammation in order to be successful in TBI. While astroglial and microglial activation is evident in the brain of TBI mice, oral administration of GTB reduces the number of activated astroglia and microglia in TBI mice [11]. GTB treatment also decreases the level of iNOS protein in the brain and reduces the number of iNOS-expressing astroglia and microglia in hippocampus and cortex of TBI mice [11].
Reduction of Axonal Damage
It is established that combination of mechanical insult and resultant neuroinflammation have a remarkable impact on the synaptic structure and function, leading to synaptic dysfunction [87]. It is also known that several plasticity-related proteins such as PSD-95, NR2A and GluR1 are involved in synapse development and maturation [76,88,89]. Studies show that CCI- induced TBI decreases the levels of PSD-95, NR2A and GluR1 in the hippocampus region of the TBI mice compared to sham control [11]. Interestingly, oral GTB treatment leads to upregulation and/or normalization of PSD-95, NR2A and GluR1 in the hippocampus of TBI mice [11]. Therefore, GTB treatment can evidently contribute to synapse development and maturation in the hippocampus of TBI mice.
Decrease in Lesion Volume
Lesion volume is an important measure used to evaluate the degree and severity of injury in TBI brains. The Cavalieri stereological technique is used to determine the lesion volume in rodents with TBI [11,12,90]. In a study with CCI-induced TBI in mice, a typical lesion with distend cavity originating from cortex through hippocampus and involving to the lateral ventricle is seen. However, daily oral treatment with GTB leads to decrease in total lesion volume in the whole hemisphere of TBI mice as compared to untreated TBI mice [11]. Therefore, GTB is capable of reducing lesion volume in TBI mice.
Improvement of Cognitive Functions
Cognitive deficiency is common among many TBI survivors and it has been reported that the cognitive defects in TBI is probably due to impaired synaptic alterations [91,92]. Since GTB treatment attenuates neuroinflammation, decreases lesion volume and restores synaptic maturation in TBI mice, the study also investigates the effect of GTB on cognitive functions and reports significant improvement in spatial memory and learning and short- term memory in TBI mice after GTB treatment [11]. Therefore, this flavoring agent may be considered for improving cognitive functions.
Restoration of Locomotor Activities
In many individuals, TBI can disturb balance and coordination, thus increasing the risk of falls, especially in the elderly. Therefore, improving locomotor function after a TBI may have a remarkable influence on recovery journey and overall well-being of a patient. It has been reported that treatment of CCI-induced TBI mice with oral GTB leads to significant increase in distance travelled, velocity, center frequency, rearing, rotarod performance, beam walking, and grid runway as compared to untreated TBI mice [11]. Therefore, oral GTB is capable of restoring and/or improving open field behavior, motor coordination and balance activity, and gait behavior in a mouse model of TBI.
Conclusion
Currently, no therapies are available that can certainly halt the progression of TBI. Although some treatments are there for taking care of blood clots, muscle spasms, anxiety, depression, and mood swings, many of the available drugs simply display symptomatic support. Moreover, the available drugs exhibit a number of undesirable effects. Therefore, development of effective treatment options for TBI is an important area of research. There are various advantages of NaB and GTB over available anti-TBI therapies. First, NaB has therapeutical importance in treating urea cycle disorders and glycine encephalopathy. GTB is also an FDA- permitted flavoring constituent for being used in food and food packaging businesses. Second, both NaB and GTB can be taken orally, the least painful route of drug treatment. Consistent with that found in mouse models MS, PD, AD, and Lewy body dementia, NaB reduces glial activation in the hippocampus and protects memory and learning in TBI mice [57,83-85]. Neuroprotective effects of GTB have also been reported in mouse models of Huntington disease and multiple sclerosis and a monkey model of Parkinson’s disease [93-95]. Accordingly, oral GTB also exhibits beneficial effects in TBI mice [11]. Therefore, these benzoate drugs (NaB and GTB) may be considered for repurposing as therapeutic agents in TBI treatment.
Data Availability
This is a review and the readers can access all the published article supporting the conclusions of this study through PubMed.
Conflicts of Interest
None
Acknowledgements
This study was supported by merit awards (1I01BX005002 and 1I01BX005613) from US Department of Veterans Affairs and a grant from NIH (AT10980). Moreover, Dr. Pahan is the recipient of a Research Career Scientist Award (1IK6 BX004982) from the Department of Veterans Affairs. However, the views expressed in this article are those of the authors and do not necessarily reflect the position or policy of the Department of Veterans Affairs or the United States government.
References
- Loane, D. J., & Faden, A. I. (2010). Neuroprotection for traumatic brain injury: translational challenges and emerging therapeutic strategies. Trends in pharmacological sciences, 31(12), 596-604.
- Maas, A. I., Stocchetti, N., & Bullock, R. (2008). Moderate and severe traumatic brain injury in adults. The Lancet Neurology, 7(8), 728-741.
- Miller, G. F., Kegler, S. R., & Stone, D. M. (2020). Traumatic brain injury-related deaths from firearm suicide: United States, 2008-2017. American journal of public health, 110(6), 897-899.
- Giner, J., Galán, L. M., Teruel, S. Y., Espallargas, M. G., López, C. P., Guerrero, A. I., & Frade, J. R. (2022). Traumatic brain injury in the new millennium: new population and new management. Neurología (English Edition), 37(5), 383-389.
- Simon, D. W., McGeachy, M. J., Bayir, H., Clark, R. S., Loane,D. J., & Kochanek, P. M. (2017). The far-reaching scope of neuroinflammation after traumatic brain injury. Nature Reviews Neurology, 13(3), 171-191.
- Blennow, K., Brody, D. L., Kochanek, P. M., Levin, H., McKee, A., Ribbers, G. M., ... & Zetterberg, H. (2016). Traumatic brain injuries. Nature reviews Disease primers, 2(1), 16084.
- Khellaf, A., Khan, D. Z., & Helmy, A. (2019). Recent advances in traumatic brain injury. Journal of neurology, 266(11), 2878- 2889.
- Poddar, J., Rangasamy, S. B., & Pahan, K. (2024). Therapeutic efficacy of cinnamein, a component of balsam of Tolu/Peru, in controlled cortical impact mouse model of TBI. Neurochemistry international, 176, 105742.
- Rangasamy, S. B., Ghosh, S., & Pahan, K. (2020). RNS60, a physically-modified saline, inhibits glial activation, suppresses neuronal apoptosis and protects memory in a mouse model of traumatic brain injury. Experimental Neurology, 328, 113279.
- Xiong, Y., Mahmood, A., & Chopp, M. (2013). Animal models of traumatic brain injury. Nature Reviews Neuroscience, 14(2), 128-142.
- Rangasamy, S. B., Poddar, J., & Pahan, K. (2023). Protection of mice from controlled cortical impact injury by food additive glyceryl tribenzoate. International Journal of Molecular Sciences, 24(3), 2083.
- Rangasamy, S. B., Raha, S., Dasarathy, S., & Pahan, K. (2021). RETRACTED: Sodium Benzoate, a Metabolite of Cinnamon and a Food Additive, Improves Cognitive Functions in Mice after Controlled Cortical Impact Injury. International journal of molecular sciences, 23(1), 192.
- Helmick, K. M., Spells, C. A., Malik, S. Z., Davies, C. A., Marion, D. W., & Hinds, S. R. (2015). Traumatic brain injury in the US military: Epidemiology and key clinical and research programs. Brain Imaging and Behavior, 9(3), 358-366.
- Howlett, J. R., Nelson, L. D., & Stein, M. B. (2022). Mental health consequences of traumatic brain injury. Biological Psychiatry, 91(5), 413-420
- Roy, A., & Pahan, K. (2015). PPARα signaling in the hippocampus: Crosstalk between fat and memory. Journal of Neuroimmune Pharmacology, 10(1), 30-34.
- Ramos-Cejudo, J., Wisniewski, T., Marmar, C., Zetterberg, H., Blennow, K., de Leon, M. J., & Fossati, S. (2018). Traumatic brain injury and Alzheimer's disease: The cerebrovascular link. EBioMedicine, 28, 21-30.
- Taylor, C. A., & Jung, H. Y. (1998). Disorders of mood after traumatic brain injury. Seminars in Clinical Neuropsychiatry, 3(3), 224-231.
- Jorge, R. E., & Arciniegas, D. B. (2014). Mood disorders after TBI. Psychiatric Clinics of North America, 37(1), 13-29.
- Dehbozorgi, M., et al. (2024). Incidence of anxiety after traumatic brain injury: A systematic review and meta-analysis. BMC Neurology, 24(1), 293.
- Vasterling, J. J., Jacob, S. N., & Rasmusson, A. (2018). Traumatic brain injury and posttraumatic stress disorder: Conceptual, diagnostic, and therapeutic considerations in the context of co-occurrence. Journal of Neuropsychiatry and Clinical Neurosciences, 30(2), 91-100.
- Loignon, A., Ouellet, M. C., & Belleville, G. (2020). A systematic review and meta-analysis on PTSD following TBI among military/veteran and civilian populations. Journal of Head Trauma Rehabilitation, 35(1), E21-E35.
- Dever, A., Powell, D., Graham, L., Mason, R., Das, J., Marshall, S. J., ... & Stuart, S. (2022). Gait impairment in traumatic brain injury: A systematic review. Sensors, 22(4), 1480.
- Delic, V., Beck, K. D., Pang, K. C. H., & Citron, B. A. (2020). Biological links between traumatic brain injury and Parkinson’s disease. Acta Neuropathologica Communications, 8(1), 45.
- Bae, Y. H., Jang, Y., Kim, S., Lee, J., Park, J., Lee, J., … & Kim, J. (2018). Brain injury induces HIF-1α-dependent transcriptional activation of LRRK2 that exacerbates brain damage. Cell Death & Disease, 9(11), 1125.
- Impellizzeri, D., Campolo, M., Bruschetta, G., Crupi, R., Cordaro, M., Paterniti, I., … & Cuzzocrea, S. (2016). Traumatic brain injury leads to development of Parkinson’s disease-related pathology in mice. Frontiers in Neuroscience, 10, 458.
- Mondal, S., Brahmachari, S., Dasarathi, S., & Pahan,K. (2024). Nebulization of low-dose aspirin ameliorates Huntington's pathology in N171-82Q transgenic mice. NeuroImmune Pharmacology and Therapeutics, 3(1), 47-59.
- Raha, S., Brahmachari, S., Dasarathi, S., & Pahan, K. (2024). Cinnamic acid, a natural plant compound, exhibits neuroprotection in a mouse model of Sandhoff disease via PPARα. NeuroImmune Pharmacology and Therapeutics, 3(1), 17-32.
- Chakrabarti, S., Gorai, S., & Pahan, K. (2023). A simple protocol for isolating microglia from adult mouse brain. NeuroImmune Pharmacology and Therapeutics, 2(3), 293-300.
- Pahan, K. (2006). Lipid-lowering drugs. Cellular and Molecular Life Sciences, 63(10), 1165-1178.
- Dutta, D., Raha, S., Roy, A., & Pahan, K. (2021). Selective targeting of the TLR2/MyD88/NF-κB pathway reduces α-synuclein spreading in vitro and in vivo. Nature Communications, 12(1), 5382.
- Saha, R. N., & Pahan, K. (2006). Regulation of inducible nitric oxide synthase gene in glial cells. Antioxidants & Redox Signaling, 8(5-6), 929-947.
- Patani, R., Hardingham, G. E., & Liddelow, S. A. (2023). Functional roles of reactive astrocytes in neuroinflammation and neurodegeneration. Nature Reviews Neurology, 19(7), 395-409.
- Du, D., Xu, F., Yu, L., Zhang, C., Lu, X., Yuan, H., … &Zheng, J. (2010). The tight junction protein occludin regulates the directional migration of epithelial cells. Developmental Cell, 18(1), 52-63.
- Shi, H., Hu, X., Leak, R. K., Shi, Y., An, C., Suenaga, J., … & Gao, Y. (2015). Demyelination as a rational therapeutic target for ischemic or traumatic brain injury. Experimental Neurology, 272, 17-25.
- Jana, A., Hogan, E. L., & Pahan, K. (2009). Ceramide and neurodegeneration: Susceptibility of neurons and oligodendrocytes to cell damage and death. Journal of the Neurological Sciences, 278(1-2), 5-15.
- Jana, A., & Pahan, K. (2010). Sphingolipids in multiple sclerosis. Neuromolecular Medicine, 12(4), 351-361.
- Nave, K. A. (2010). Myelination and support of axonal integrity by glia. Nature, 468(7321), 244-252.
- Jana, M., Ghosh, S., & Pahan, K. (2018). Upregulation of myelin gene expression by a physically modified saline via phosphatidylinositol 3-kinase-mediated activation of CREB: Implications for multiple sclerosis. Neurochemical Research, 43(2), 407-419.
- Jana, M., & Pahan, K. (2013). Down-regulation of myelin gene expression in human oligodendrocytes by nitric oxide: Implications for demyelination in multiple sclerosis. Journal of Clinical & Cellular Immunology, 4, 149.
- Caprariello, A. V., Mangla, S., Miller, R. H., & Selkirk, S.M. (2012). Apoptosis of oligodendrocytes in the central nervous system results in rapid focal demyelination. Annals of Neurology, 72(3), 395-405
- Back, S.A., Craig,A., Luo, N. L., Ren, J.,Akundi, R. S., Ribeiro, I., … & Volpe, J. J. (2007). Hypoxia-ischemia preferentially triggers glutamate depletion from oligodendroglia and axons in perinatal cerebral white matter. Journal of Cerebral Blood Flow & Metabolism, 27(2), 334-347.
- Petito, C. K., Olarte, J. P., Roberts, B., Nowak, T. S., & Pulsinelli, W. A. (1998). Selective glial vulnerability following transient global ischemia in rat brain. Journal of Neuropathology & Experimental Neurology, 57(3), 231-238.
- Kang, J. H., & Lin, H. C. (2012). Increased risk of multiple sclerosis after traumatic brain injury: A nationwide population- based study. Journal of Neurotrauma, 29(1), 90-95.
- Nave, K. A., & Trapp, B. D. (2008). Axon-glial signaling and the glial support of axon function. Annual Review of Neuroscience, 31, 535-561.
- Bremer, J., Baumann, F., Tiberi, C., Wessig, C., Fischer, H., Schwarz, P., … & Nave, K. A. (2010). Axonal prion protein is required for peripheral myelin maintenance. Nature Neuroscience, 13(3), 310-318.
- Lappe-Siefke, C., Goebbels, S., Gravel, M., Nicksch, E., Lee, J., Braun, P. E., … & Nave, K. A. (2003). Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nature Genetics, 33(3), 366-374.
- Dutta, R., & Trapp, B. D. (2007). Pathogenesis of axonal and neuronal damage in multiple sclerosis. Neurology, 68(22 Suppl 3), S22-S31.
- Abouelmagd, M. E., Abd-Elrahman, M. E., Ahmed, M. M., & Abdelrahman, H. (2024). History of head trauma and the risk of multiple sclerosis: A systematic review and meta-analysis. Multiple Sclerosis and Related Disorders, 92, 106183.
- Jha, R. M., Kochanek, P. M., & Simard, J. M. (2019). Pathophysiology and treatment of cerebral edema in traumatic brain injury. Neuropharmacology, 145(Pt B), 230-246.
- Jana, M., & Pahan, K. (2005). Redox regulation of cytokine- mediated inhibition of myelin gene expression in human primary oligodendrocytes. Free Radical Biology and Medicine, 39(6), 823-831.
- Jana, A., & Pahan, K. (2007). Oxidative stress kills human primary oligodendrocytes via neutral sphingomyelinase: Implications for multiple sclerosis. Journal of Neuroimmune Pharmacology, 2(2), 184-193.
- Roy, A., Jana, A., Yatish, K., Freidt, M. B., Fung, Y. K., Martinson, J. A., & Pahan, K. (2008). Reactive oxygen species up-regulate CD11b in microglia via nitric oxide: Implications for neurodegenerative diseases. Free Radical Biology and Medicine, 45(5), 686-699.
- Barnham, K. J., Masters, C. L., & Bush, A. I. (2004). Neurodegenerative diseases and oxidative stress. Nature Reviews Drug Discovery, 3(3), 205-214.
- Fesharaki-Zadeh, A. (2022). Oxidative stress in traumatic brain injury. International Journal of Molecular Sciences, 23(21), 13000.
- Lorente, L. (2017). Biomarkers associated with the outcome of traumatic brain injury patients. Brain Sciences, 7(11), 142.
- Pahan, K., Sheikh, F. G., Namboodiri, A. M. S., & Singh,(1998). N-acetyl cysteine inhibits induction of NO production by endotoxin or cytokine-stimulated rat peritoneal macrophages, C6 glial cells, and astrocytes. Free Radical Biology and Medicine, 24(1), 39-48.
- Modi, K. K., Roy, A., Brahmachari, S., Rangasamy, S. B., & Pahan, K. (2015). Cinnamon and its metabolite sodium benzoate attenuate the activation of p21rac and protect memory and learning in an animal model of Alzheimer’s disease. PLoS ONE, 10(6), e0130398.
- Lambeth, J. D. (2004). NOX enzymes and the biology of reactive oxygen. Nature Reviews Immunology, 4(3), 181-189.
- Pahan, K. (2011). Immunomodulation of experimental allergic encephalomyelitis by cinnamon metabolite sodium benzoate. Immunopharmacology and Immunotoxicology, 33(4), 586-593.
- Pahan, P., & Pahan, K. (2015). Can cinnamon bring aroma in Parkinson's disease treatment? Neural Regeneration Research, 10(1), 30-32.
- Pahan, K. (2015). Prospects of cinnamon in multiple sclerosis.Journal of Multiple Sclerosis (Foster City), 2(3), 149.
- Pahan, S., & Pahan, K. (2020). Can cinnamon spice down autoimmune diseases? Journal of Clinical & Experimental Immunology, 5(6), 252-258.
- Corbett, G. T., Roy, A., & Pahan, K. (2012). Gemfibrozil, a lipid-lowering drug, upregulates IL-1 receptor antagonist in mouse cortical neurons: Implications for neuronal self- defense. The Journal of Immunology, 189(2), 1002-1013.
- Khasnavis, S., Roy, A., Ghoshal, N., Watson, C. M., & Pahan,K. (2013). Castration induces Parkinson disease pathologies in young male mice via inducible nitric-oxide synthase. Journal of Biological Chemistry, 288(29), 20843-20855.
- Saha, R. N., & Pahan, K. (2006). Signals for the induction of nitric oxide synthase in astrocytes. Neurochemistry International, 49(2), 154-163.
- Pahan, K., & Mondal, S. (2012). Crosstalk between Nitric Oxide and T helper cells. Journal of clinical & cellular immunology, 3(4), e109.
- Mondal, S., Brahmachari, S., & Pahan, K. (2012). Regulation of encephalitogenicity of neuroantigen-primed T cells by nitric oxide: Implications for multiple sclerosis. Journal of Clinical & Cellular Immunology, 3(3), 124.
- Brahmachari, S., Jana, A., & Pahan, K. (2009). Sodium benzoate, a metabolite of cinnamon and a food additive, reduces microglial and astroglial inflammatory responses. The Journal of Immunology, 183(9), 5917-5927.
- Iaccarino, M. A., Bhatnagar, S., & Zafonte, R. (2015). Rehabilitation after traumatic brain injury. Handbook of Clinical Neurology, 127, 411-422.
- Modi, K. K., Rangasamy, S. B., Dasarathy, S., Roy, A., & Pahan, K. (2016). Cinnamon converts poor learning mice to good learners: Implications for memory improvement. Journal of Neuroimmune Pharmacology, 11(4), 693-707.
- Patel, D., Roy, A., Kundu, M., Jana, M., Luan, C. H., Gonzalez, F. J., & Pahan, K. (2020). Upregulation of BDNF and hippocampal functions by a hippocampal ligand of PPARα. JCI Insight, 5(10), e136654.
- Rangasamy, S. B., Roy, A., Dasarathy, S., & Pahan, K. (2023). Treadmill workout activates PPARα in the hippocampus to upregulate ADAM10, decrease plaques and improve cognitive functions in 5XFAD mouse model of Alzheimer's disease. Brain, Behavior, and Immunity, 109, 204-218.
- Leng, F., & Edison, P. (2021). Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nature Reviews Neurology, 17(3), 157-172.
- Korczyn, A. D., & Grinberg, L. T. (2024). Is Alzheimer disease a disease? Nature Reviews Neurology, 20(4), 245-251.
- Chandra, S., Roy, A., Jana, M., Kundu, M., & Pahan, K. (2019). PPARα between aspirin and plaque clearance. Journal of Alzheimer’s Disease, 71(2), 389-397.
- Patel, D., Roy, A., Kundu, M., Jana, M., Luan, C. H., Gonzalez,F. J., & Pahan, K. (2018). Aspirin binds to PPARα to stimulate hippocampal plasticity and protect memory. Proceedings of the National Academy of Sciences of the United States of America, 115(31), E7408-E7417.
- Dutta, D., Jana, M., Majumder, M., Roy,A., & Pahan, K. (2023). Tau fibrils induce glial inflammation and neuropathology viaTLR2 in Alzheimer's disease-related mouse models. Journal of Clinical Investigation, 133(18), e167748.
- Corbett, G. T., Gonzalez, F. J., & Pahan, K. (2015). Activation of peroxisome proliferator-activated receptor α stimulates ADAM10-mediated proteolysis of APP. Proceedings of the National Academy of Sciences of the United States of America, 112(27), 8445-8450.
- Johnson, V. E., Stewart, W., & Smith, D. H. (2010). Traumatic brain injury and amyloid-β pathology: A link to Alzheimer's disease? Nature Reviews Neuroscience, 11(5), 361-370.
- Rangasamy, S. B., Roy, A., Dasarathy, S., & Pahan, K. (2019). Low-dose aspirin upregulates tyrosine hydroxylase and increases dopamine production in dopaminergic neurons: Implications for Parkinson's disease. Journal of Neuroimmune Pharmacology, 14(2), 173-187.
- Lan, Y. L., Chen, J. J., Hu, G., Xu, J., Xiao, M., Li, S., & Huang,Y. (2019). The potential roles of dopamine in traumatic brain injury: A preclinical and clinical update. American Journal of Translational Research, 11(5), 2616-2631.
- Rangasamy, S. B., Roy, A., Dasarathy, S., & Pahan, K. (2021). Stimulation of dopamine production by sodium benzoate, a metabolite of cinnamon and a food additive. Journal of Alzheimer’s Disease Reports, 5(1), 295-310.
- Khasnavis, S., & Pahan, K. (2014). Cinnamon treatment upregulates neuroprotective proteins Parkin and DJ-1 and protects dopaminergic neurons in a mouse model of Parkinson's disease. Journal of Neuroimmune Pharmacology, 9(4), 569-581.
- Brahmachari, S., & Pahan, K. (2007). Sodium benzoate, a food additive and a metabolite of cinnamon, modifies T cells at multiple steps and inhibits adoptive transfer of experimental allergic encephalomyelitis. The Journal of Immunology, 179(1), 275-283.
- Raha, S., Roy, A., Dasarathy, S., Rangasamy, S. B., & Pahan,K. (2021). Reduction of Lewy body pathology by oral cinnamon. Journal of Neuroimmune Pharmacology, 16(3), 592-608.
- Pahan, S., Dasarathi, S., & Pahan, K. (2021). Glyceryl tribenzoate: A food additive with unique properties to be a substitute for cinnamon. Journal of Clinical & Experimental Immunology, 6(5), 367-372.
- Sweeney, N., Zhang, Y., Liu, Y., & others. (2024). Neuronal BAG3 attenuates tau hyperphosphorylation, synaptic dysfunction, and cognitive deficits induced by traumatic brain injury via the regulation of autophagy-lysosome pathway. Acta Neuropathologica, 148(1), 52.
- Roy, A., Jana, M., Corbett, G. T., Ramaswamy, S., Kordower,J. H., Gonzalez, F. J., & Pahan, K. (2013). Regulation of cyclic AMP response element binding and hippocampal plasticity- related genes by peroxisome proliferator-activated receptor α. Cell Reports, 4(4), 724-737.
- Roy, A., Ghosh, A., Jana, M., Liu, X., Brahmachari, S., Gendelman, H. E., & Pahan, K. (2014). Enhancement of morphological plasticity in hippocampal neurons by a physically modified saline via phosphatidylinositol-3 kinase. PLoS ONE, 9(7), e101883.
- Dhillon, N. K., Rappaport, M., & others. (2020). How repetitive traumatic injury alters long-term brain function. Journal of Trauma and Acute Care Surgery, 89(5), 955-961.
- Alashram, A. R., Annino, G., Padua, E., & Romagnoli, C. (2019). Cognitive rehabilitation post-traumatic brain injury: A systematic review for emerging use of virtual reality technology. Journal of Clinical Neuroscience, 66, 209-219.
- Fronczak, K. M., Johnson, E. K., & others. (2021). Reductions in synaptic vesicle glycoprotein 2 isoforms in the cortex and hippocampus in a rat model of traumatic brain injury. Molecular Neurobiology, 58(11), 6006-6019.
- Dutta, D., Roy, A., Rangasamy, S. B., Dasarathy, S., & Pahan,K. (2021). Alleviation of Huntington pathology in mice by oral administration of food additive glyceryl tribenzoate. Neurobiology of Disease, 153, 105318.
- Mondal, S., Dasarathi, S., & Pahan, K. (2017). Glyceryl tribenzoate: A flavoring ingredient, inhibits the adoptive transfer of experimental allergic encephalomyelitis via TGF-β: Implications for multiple sclerosis therapy. Journal of Clinical & Cellular Immunology, 8(1), 1000489.
- Rangasamy, S. B., Roy, A., Dasarathy, S., & Pahan, K. (2022). Protection of dopaminergic neurons in hemiparkinsonian monkeys by flavouring ingredient glyceryl tribenzoate. NeuroImmune Pharmacology and Therapeutics, 1(1), 7-22.