

Research Article - (2026) Volume 9, Issue 2
An Unusual Case of Creutzfeldt-Jakob Disease Misdiagnosed as Status Epilepticus: A Case Study
2MGM Medical College, Aurangabad, India
Received Date: Mar 06, 2026 / Accepted Date: Mar 30, 2026 / Published Date: Apr 09, 2026
Copyright: ©2026 Singh Karminder, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Citation: Karminder, S., Niraj, A., Kakde, S., Chandershekran, P. N., Rameez, M. (2026). An Unusual Case of Creutzfeldt- Jakob Disease Misdiagnosed as Status Epilepticus: A Case Study. Adv Neur Neur Sci, 9(2), 01-09.
Abstract
Introduction: Creutzfeldt-Jakob disease (CJD) is a rare neurodegenerative disease worldwide, with approximately 1–1.5 cases per million people per year. It is caused by a type of abnormal protein known as a prion or scrapie PrP. Infectious prions are misfolded proteins that can induce misfolding in normally folded proteins, leading to clinical symptoms. Clinical symptoms vary, with early symptoms including memory problems, behavioral changes, poor coordination, and visual and auditory disturbances. Later symptoms include dementia, involuntary movements, blindness, deafness, weakness, and coma. The varied clinical presentations pose diagnostic challenges for clinicians, requiring multiple diagnostic modalities and a high index of suspicion.
Case Presentation: We present a case of a 54-year-old man with a history of complex partial seizures, diabetes mellitus type 2, left temporal lobe surgery secondary to seizures, deep vein thrombosis and pulmonary embolism, left common to external iliac vein wall stent, hypertension, and hyperlipidemia, who was admitted to the Neuro ICU after a motor vehicle accident and was found to have seizures at the time of extrication. He was intubated at the site due to the inability to protect the airway with concern for status epilepticus. The patient had a protracted ICU course due to a lack of improvement despite being treated with multiple antiseizure medications and a negative workup. Furthermore, a rapidly progressive neurological decline marked by refractory encephalopathy, abnormal involuntary movements, and evolving focal-to-diffuse neuroimaging abnormalities developed. Ultimately, the patient tested positive for 14-3-3 in the CSF and was diagnosed with Creutzfeldt- Jakob Disease (CJD) as a delayed diagnosis.
Conclusion: Creutzfeldt-Jakob disease (CJD) is a rare disease with varied presentations, posing diagnostic challenges for clinicians. We highlight an interesting case of Creutzfeldt- Jakob Disease (CJD), which was misdiagnosed as status epilepticus due to an unusual presentation causing a diagnostic dilemma. The case highlights features of the clinical examination and imaging findings that are atypical of CJD. Clinicians should be vigilant in the management of patients with status epilepticus who do not respond to conventional antiepileptic medications and may have an alternative diagnosis.
Keywords
Creutzfeldt-Jakob Disease (CJD), Prion Disease, Status Epilepticus, Unusual Presentation, MisdiagnosisBackground
Creutzfeldt-Jakob Disease (CJD) is a sporadic neurodegenerative disease worldwide caused by prions—proteinaceous infectious particles composed of misfolded, β-sheet–rich, protease-resistant isoforms of the normal cellular prion protein (PrPC), termed PrPSC, which lack nucleic acids and propagate by inducing conformational conversion of native proteins [1-3]. The name "Creutzfeldt–Jakob disease" was introduced by Walther Spielmeyer in 1922, after the German neurologists Hans Gerhard Creutzfeldt and Alfons Maria Jakob [4]. Creutzfeldt-Jakob disease (CJD) is a rare, rapidly progressing, invariably fatal disease that belongs to a family of diseases known as prion diseases, also called transmissible spongiform encephalopathies (TSE). In addition to abnormal prion protein accumulation in the brain, CJD is characterized by spongiform changes, neuronal loss, and gliosis [5]. Clinical symptoms vary, with early symptoms including memory problems, behavioral changes, poor coordination, visual disturbances, and auditory disturbances. Later symptoms include dementia, involuntary movements, blindness, deafness, weakness, and coma [6].
The incidence of CJD worldwide is one to two cases per million individuals per year. In the United States, these statistics translate to nearly 500 new cases per year, with one CJD death per every 6000–10000 deaths in the U.S. each year [7]. The mean age of onset is 62 years, although it has also been reported in younger and older age groups [8,9] . Sporadic CJD has a 1:1 male-to-female ratio. Death occurs in nearly 70% of patients within a year of onset. The mean survival of sporadic CJD is 4–8 months, with 90% of patients dying within a year.
There are various subtypes of CJD, but sporadic Creutzfeldt-Jakob disease (sCJD) is the most common in humans (85-90%), while the rest are iatrogenic, familial, and variant forms [10]. The main symptoms of CJD are cognitive decline, leading to encephalopathy, dementia, myoclonus, and lack of coordination of movement. Diagnosing CJD is challenging for physicians because it is a rare disease with diverse symptoms. Key diagnostic tools for CJD include electroencephalography (EEG), MRI, and CSF studies. CSF with Real-Time Quaking-Induced Conversion (RT-QuIC) or olfactory mucosa samples have high sensitivity (96%) and almost 100% specificity, and tests for 14-3-3 and Tau proteins provide excellent diagnostic value [11]. We present a case of a male who presented to the University Hospital with seizures but was later found to have unusual clinical features and changes in brain imaging, which prompted us to consider further diagnostic testing, which revealed CJD.
Case Presentation
A 54-year-old man with a history of complex partial seizures, diabetes mellitus type 2, left temporal lobe surgery secondary to seizures, deep vein thrombosis, pulmonary embolism, left common to external iliac vein wall stent, hypertension, and hyperlipidemia, was witnessed to have a generalized tonic-clonic seizure after a motor vehicle accident. He was intubated at the site because he was unable to protect his airway. Upon arrival at the Emergency Department, the patient continued to have tonic-clonic seizure activity and was loaded with Levitricetam 4000 mg IV, Fosphenytoin 1600 mg phenytoin equivalent (PE) IV, and 8 mg of Midazolam, but continued to have generalized tonic-clonic seizures for which he was given Lacosamide 200 mg IV. Admission to the Neurosciences Intensive Care Unit (NSICU) was considered for further management.
The patient remained critically ill in the Neurosciences Intensive Care Unit (NSICU) and was placed on mechanical ventilation with propofol as a sedative agent. Subsequently, initiated on levetiracetam 1500 mg IV twice daily and carbamazepine 400 mg IV twice daily as maintenance doses. Due to concerns of aspiration, piperacillin-tazobactam 4.5 grams IV every 8 hours was commenced empirically. The initial investigations included blood analysis, with an elevated white blood cell count of 18.80 × 10 (9)/L, hemoglobin of 13.9 g/dL, and platelets of 317 × 10 (9)/L. He also had elevated lactic acid levels (4.0 mg/dl), which later improved during the ICU stay. Chest radiography revealed no acute cardiopulmonary findings. Trauma workup, including a computed tomography (CT) scan of the head, showed no acute parenchymal or extra-axial hemorrhage but revealed left anterior temporal lobe chronic encephalomalacia and advanced hippocampal atrophy, which was consistent with a history of prior left temporal lobe surgery (due to seizures). Abdominal CT showed minimal air in the soft tissues of the right anterior lower abdomen, possibly representing a laceration or injection site. No acute posttraumatic intrathoracic, abdominal, or pelvic findings were observed, except for bilateral iliac vein stents with probable thrombus, similar to the previous CT venogram. A CT angiogram of the chest was positive for a probable right lower lobe chronic pulmonary embolus. Continuous video electroencephalography (cEEG) was initiated on the day of admission. Electroencephalography (EEG) on day 1 showed diffuse slowing of the background, with 2-5 Hz theta and delta activity, high-amplitude focal slowing, and almost continuous focal periodic epileptiform abnormalities in the right temporoparietal region.
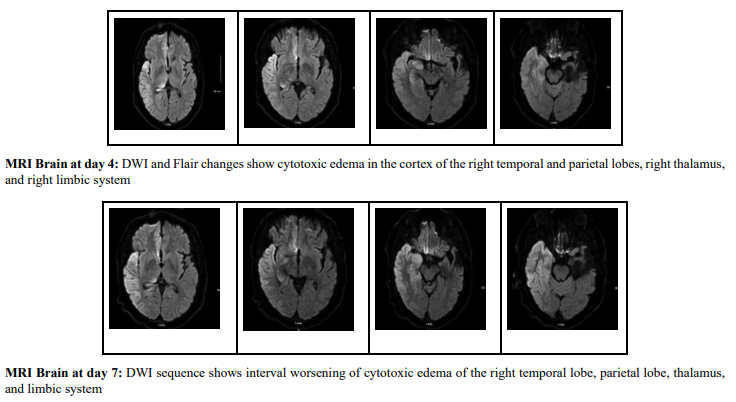
Remained intubated on day 3, but on weaning the sedation, he was found to have left facial twitching. EEG correlated with focal electrographic seizures and was associated with focal twitching in the face concerning focal motor seizures. MRI of the brain (Day 4) was performed because of the inability to move the left side of the body during sedation reduction and showed cytotoxic edema in the right temporal and parietal cortex, ipsilateral limbic system, and right thalamus. Left anterior temporal lobe chronic encephalomalacia with advanced hippocampal atrophy. Due to the history of pulmonary embolism, MR Venogram was performed to rule out cerebral venous thrombosis, which was found to be negative. Even though the patient’s clinical condition remained the same, the EEG showed mild diffuse encephalopathy and a superimposed right hemispheric cortical and subcortical dysfunction over the left temporal region, with no discharges or seizures on day 5. At this time, we discontinued the EEG. Propofol was changed to dexmedetomidine as the patient became agitated during weaning sedation.
On day 7, the patient again had abnormal involuntary movements involving the jaw and tongue, which were exacerbated after adding quetiapine because of his persistent agitation. A presumed diagnosis of Tardive Dyskinesia (TD) was made due to worsening abnormal involuntary movements after quetiapine administration. We discontinued quetiapine, added hydroxyzine for agitation, and restarted continuous EEG monitoring. Again, dexmedetomidine was administered due to persistent agitation despite the administration of clonazepam and hydroxyzine. EEG on day 10 showed mild to moderate diffuse encephalopathy and superimposed right hemispheric cortical and subcortical dysfunction, in addition to evidence of a skull defect over the left temporal region and potential epileptogenicity from the left temporal head region. Although bilateral facial twitching events, more on the left than on the right, were not associated with epileptiform or rhythmic abnormalities. The patient continued to experience facial twitching despite being on multiple antiepileptic medications.
Unfortunately, on day 12, the patient deteriorated with hypoxic respiratory failure and was unable to manage oral secretions, resulting in reintubation and placement on mechanical ventilation. Antibiotics were restarted due to concerns of hospital-acquired pneumonia due to worsening leukocytosis and new opacities on chest x-ray. Physical examination revealed neck stiffness with positive Kernig’s and Brudzinski’s signs, in addition to leukocytosis and worsening mentation. The decision to perform a lumbar puncture was delayed because the patient was on Clopidogrel and Apixaban, which were held. Vancomycin, Ceftriaxone, Acyclovir, and Ampicillin were started as empiric treatment for meningitis-encephalitis.
On day 12, MRI brain showed interval worsening of cytotoxic edema of the right temporal lobe, parietal lobe, thalamus, and limbic system. Lumbar puncture was performed on day 14, with preliminary results showing proteins of 39 mg/dL, glucose of 113 mg/dL, and white cell count of 1 /mcL. Quantitative tracheal culture (QTL) grew staphylococcus aureus, which was later identified as methicillin-sensitive staphylococcus aureus (MSSA). As CSF studies did not indicate bacterial meningitis, antibiotics were discontinued, and no accurate diagnosis was made at that time. The patient remained intubated and failed the breathing trials. The family was informed about the potential tracheostomy and percutaneous endoscopic gastrostomy (PEG). At this time, a presumed diagnosis of autoimmune encephalitis was made, and CSF studies were sent and started on five days of intravenous high-dose steroids (1 gram of methylprednisolone). No significant improvement was noted after 5 days of intravenous steroids, as the patient still had dyskinesias involving the tongue and jaw.
On day 19, he was on three anti-epileptic medications, including Lacosamide, Carbamazepine, and Brivaracetam. CSF studies revealed a negative antibody panel for autoimmune encephalitis (AE). Repeat MRI of the brain on day 20, remarkable for increased bilateral cerebral hemispheric diffuse cortical and deep nuclei cytotoxic edema. Due to the high suspicion of autoimmune encephalitis as a differential diagnosis, a workup was performed to rule out underlying cancer with a negative CT chest, abdomen, and pelvis. On day 21, a repeat lumbar puncture showed a white cell count of 0 and a protein level of 42 mg/dl. CSF was also sent for prion marker analysis. Owing to the lack of improvement, a decision was made to start plasmapheresis (PLEX) on day 22 and continue for a total of five sessions. To date, we have exhausted all treatment modalities, including anti-epileptics, steroids, and plasmapheresis, with no significant improvement. After a family discussion, the surgical team was consulted, and the patient underwent tracheostomy and percutaneous gastrostomy (PEG) placement on day 30. Eventually, he was discharged to a long-term acute care facility (LTAC) on day 40 of his stay in the Neuro ICU. Four days after discharge, CSF results were finalized with negative prion markers (RT-QuIC), but elevated total Tau proteins of more than 20000pg/ml (Reference Interval: 0- 1149 pg/mL) and 14-3-3 gamma level of 137669 AU/ml (Reference Interval: 173-1999 AU/ ml). The patient's family was informed of the CSF findings and advised to follow up with the neurology clinic.
|
Hospital day |
Key clinicalevents |
EEG findings |
MRI Brain findings |
Diagnostic implication |
|
0–1 |
Post-motor vehicle collision, convulsive seizures; intubated; loaded with multiple anti-seizure medications (ASMs) |
Diffuse slowing; focal periodic epileptiform abnormalities (right temporoparietal) |
— |
Strongly supports status epilepticus in an epileptic patient |
|
4 |
Left facial twitching on sedation wean |
Focal electrographic seizures correlating with twitching |
DWI: right temporo- parietal cortical + thalamic/limbic cytotoxic edema |
Can be peri-ictal; prion still possible but less suspected |
|
5 |
Extubated; seizures less evident |
Encephalopathy, no ongoing seizures/ discharges |
— |
If persistent decline continues, broaden differential |
|
7–10 |
Jaw/tongue movements; agitation; suspected TD after quetiapine |
Facial twitching events later without ictal correlate |
— |
Movement disorder not fully explained by epilepsy |
|
12–14 |
Respiratory decline; re-intubated; meningeal signs; empiric antimicrobials; Spinal tap delayed due to being on antithrombotics |
— |
DWI: persistent right hemispheric diffusion Restriction with no ADC correlate |
Progressive DWI argues against pure peri-ictal injury |
|
14 |
Spinal tap obtained |
— |
— |
CSF non inflammatory reduces infectious likelihood |
|
19–22 |
No improvement; autoimmune encephalitis suspected; steroids then Plasmapheresis (PLEX ) |
Encephalopathy; no ictal correlate for movements |
MRI day 20: diffuse bilateral cortical/deep nuclei restriction |
The pattern strongly favors prion-like destructive process |
|
30–40 |
Tracheostomy and Percutaneous Gastrostomy; discharge to LTAC |
— |
— |
Advanced neurological decline |
|
Post- discharge |
RT-QuIC negative; tau >20,000 pg/mL; 14-3-3 strongly positive |
— |
— |
Biomarkers strongly support prion-associated Rapidly progressive type (RPD) phenotype |
Table 1: Patient's Timeline in ICU
DWI: Diffusion-weighted imaging, PLEX: Plasmapheresis, LTAC: Long Term Acute Care, ADC: Apparent Diffusion Coefficient
|
Feature |
Peri-ictal injury |
Findings in our patient |
Interpretation |
|
MRI diffusion restriction |
Typically transient; improves with seizure control |
Progressive worsening and bilateral spread on serial MRI of the brain |
Favour prion-associated neurodegeneration |
|
EEG correlation |
Clinical movements usually have ictal correlate |
Persistent movements without an EEG correlate later in the course |
Suggests non epileptic movement disorder |
|
CSF profile |
Usually non-specific |
Non-inflammatory CSF with extreme tau and positive 14-3-3 |
Supports rapid neuronal injury |
|
Response to therapy |
Improvement with seizure control |
Continued deterioration despite seizure control and immunotherapy |
Inconsistent with peri-ictal process |
Table 2: Key Features Distinguishing Peri-Ictal Injury from Prion Disease in our Patient
EEG: Electroencephalography
|
CSF test |
Patient result |
What it means in context |
|
WBC |
0–1 cells/µL |
Non-inflammatory CSF supports noninfectious, noninflammatory encephalopathy |
|
14-3-3 Protein |
>137669 AU/ml |
Very high level, supports extensive neuronal damage; helpful when paired with MRI progression |
|
Total Tau proteins |
>20,000 pg/mL |
Extreme elevation strongly supports rapid neuronal injury; very high tau (>10,000) has strong diagnostic weight in sCJD cohorts |
|
RT-QuIC |
Negative |
Highly specific when positive; sensitivity <100% → false negatives occur; interpret with phenotype + MRI + tau/14-3-3 |
Table 3: Interpreting Prion Biomarkers in our Patient
Discussion
CJD is generally characterized by rapid progression and a typical survival of 4-6 months from the diagnosis to death [12]. Prions, also known as proteinaceous infectious particles, are self-propagating proteins that primarily consist of proteinase K-resistant β-pleated sheet aggregates and lack nucleic acids. These prion particles are responsible for CJD and other transmissible spongiform encephalopathies, including bovine spongiform encephalopathy, also called mad cow disease, kuru, and scrapie [13]. It has a varied clinical presentation with early symptoms of poor coordination, confusion, disorientation, delusions, behavioral changes, such as depression, mood swings, and anxiety, speech difficulty, insomnia, or changes in sleeping patterns, hallucinations, and dizziness. Late symptoms may include blindness, involuntary movements, weakness of extremities, and ultimately coma [10].
|
|
Nystagmus Ataxia |
|
Hypokinesia Bradykinesia Dystonia Rigidity |
Table 4: Symptoms of Creutzfeldt-Jakob Disease
CJD is classified as sporadic in a person with no known cause, accounting for up to 85% of cases, and is due to misfolding of normal PrP isoforms with no apparent triggers [14-17]. The subtypes of sporadic CJD include sporadic fatal insomnia and variably protease-sensitive prionopathy. Symptoms of the sporadic form usually appear between 60 and 70 years of age. The second most common type of CJD is the genetic type, which accounts for approximately 10-15%, and is due to a heritable genetic mutation. The subtypes of this condition include familial CJD, fatal familial insomnia, and Gerstmann-Sträussler-Scheinker syndrome. Hereditary CJD typically affects people at a younger age than sporadic CJD, generally under the age of 55 years. In rare cases, this can happen in the early 20s [13]. The third form of CJD is the acquired type (1%), which is transmitted by airborne or contact routes. Certain medical procedures include handling the dura mater, corneal transplantation, and implanting electrodes.
Diagnosing CJD is very challenging. Due to advanced diagnostics and reduced autopsy rates, most diagnoses are made antemortem [19,20]. The diagnostic criteria used by the International CJD Surveillance Network have evolved with advances in investigation, first adding electroencephalography (EEG) and later including cerebrospinal fluid (CSF) 14-3-3 protein measurements. Subsequently, basal ganglia hyperintensities on MRI were incorporated. The criteria were revised in 2017 and comprise multifocal cortical signal changes (i.e., ribboning) on brain MRI and the real-time quaking-induced conversion (RT-QuIC) assay, which has increased sensitivity to 97% and specificity to 99% [21]. MRI, EEG, and cerebrospinal fluid (CSF) evaluation are cornerstone for the diagnosis of patients suspected of CJD. MRI is most helpful in diagnosing CJD among neuroimaging studies [18]. The most common findings on MRI are hyperintense signals on DWI, FLAIR, and T2-weighted images involving the cerebral cortex, corpus striatum, caudate head, and putamen [30,31]. Among all sequences, DWI is the most sensitive for diagnosing CJD, especially for cortical and striatal changes [21]. Different phenotypic variants of sporadic CJD have characteristic radiographic features, including Brownell-Oppenheimer, which initially involves the cerebellum and, in some cases, the basal ganglia. The Heidenhain variant has a predilection for the parieto-occipital cortex, and the Stern-Garcin variant involves the basal ganglia and thalamus [22,23].
In our case, MRI of the brain showed changes in the right frontotemporal area on DWI sequences, which subsequently worsened, with involvement of the right caudate nucleus on the final MRI. The subsequent progression with involvement of the right caudate nucleus and worsening diffusion abnormalities on serial imaging strongly favored a neurodegenerative prion process. Importantly, peri-ictal MRI changes typically resolve or stabilize over time, whereas progressive and spreading diffusion restriction is a hallmark of sCJD [41,42]. MRI changes can be non-specific and can be found in conditions such as stroke, vasculitis, reversible posterior leukoencephalopathy, autoimmune encephalitis, hypoxic/anoxic brain injury, osmotic demyelination, hepatic encephalopathy, and mitochondrial diseases [24-26].
<img src="https://www.opastpublishers.com/scholarly-images/10544-69d8c3d66fa5c-an-unusual-case-of-creutzfeldtjakob-disease-misdiagnosed-as-.png" width="600" height="350">
<img src="https://www.opastpublishers.com/scholarly-images/10544-69d8c41329a1a-an-unusual-case-of-creutzfeldtjakob-disease-misdiagnosed-as-.png" width="600" height="180">
|
Feature |
Expected in peri ictal/post-ictal injury |
Observed in this patient |
Interpretation |
|
Course of DWI restriction |
Often transient; tends to improve with seizure control |
Worsened on day 12; became bilateral diffuse by day 20 |
Progressive spread supports prion-like process |
|
Distribution |
Can be unilateral, cortical/ hippocampal; may involve the basal ganglia rarely |
Cortex + thalamus/ limbic early; later bilateral cortex + deep nuclei |
Deep nuclei + progressive bilateral cortical ribboning favors the CJD pattern |
|
Clinical recovery |
Neuro status typically improves as seizures resolve |
Persistent decline despite an antiseizure regimen and periods without ictal EEG |
Suggests non epileptic neurodegeneration |
|
Movement phenomena |
Usually resolves with seizure control |
Persistent jaw/tongue dyskinesias and twitching without ictal EEG |
Supports encephalopathy related movement disorder |
Table 5: Why “Post-Ictal MRI” Became Less Likely in this Patient
Early-stage EEG findings range from diffuse slowing and frontal rhythmic delta activity (FIRDA) to periodic synchronous bi- or triphasic periodic sharp wave complexes (PSWC), which are highly characteristic and observed in 67-95% of patients with sporadic CJD [27]. A typical PSWC pattern involves generalized and/or lateralized complexes, periodic cerebral potentials with a duration of 100-600 milliseconds, an intercomplex interval of 500-2000 milliseconds, and at least five repetitive intervals with a duration difference of < 500 ms. These findings are suggestive but not definitive, limiting the role of the EEG as a diagnostic tool for CJD. False-positive EEG results have been reported in Alzheimer's disease and vascular dementia. In our patient, the EEG did not show any features of CJD, as described above. The EEG pattern showed diffuse slowing of background consisting of 2-5 Hz theta and delta activity, high-amplitude focal slowing along with almost continuous focal periodic epileptiform abnormalities in the right temporoparietal region on day 3, which did not change on day 10th, mild to moderate diffuse encephalopathy, and a superimposed right hemispheric cortical and subcortical dysfunction in addition to evidence of a skull defect over the left temporal region and potential epileptogenicity from the left temporal head region. Continuous monitoring of serial EEG changes can increase the diagnostic yield in CJD, as progression from non-specific changes to definitive patterns such as PSWCs [28].
CSF evaluation is necessary for the diagnosis of CJD, which includes real-time quaking-induced conversion (RT-QuIC) assay, 14-3-3 protein, and tau proteins as main markers. Disease (PrPSc)-associated prion proteins trigger a conformational change in recombinant prion protein (recPrP), leading to amyloid formation, which can be monitored in real time using the real-time quaking-induced conversion (RT-QuIC) assay [29]. 14-3-3 is a normal protein that is released into the CSF after acute neuronal injury [33]. It is an adjunctive test that has reported mixed results about the overall sensitivity of 92% and a specificity of 80% [34,35]. The importance of 14-3-3 as a biomarker for CJD has been emphasised and proposed as a diagnostic marker [36]. Few studies have suggested that 14-3-3γ could serve as a specific marker of neuronal damage in CJD, thereby enhancing its reliability in elucidating CJD pathogenesis [37,38]. Increased tau protein levels (>1150 picogram/mL) have superior accuracy and specificity for diagnosing CJD compared with 14-3-3 protein [39,40]. In one study, patients with CJD were found to have elevated CSF tau levels but not phosphorylated tau; an elevated ratio of total tau to phosphorylated tau levels had a very high specificity for CJD. CSF total tau is also a prognostic marker of survival time in sporadic CJD, whether used alone or in combination with other variables.
Our patient has a very high level of tau proteins, which increases the likelihood of CJD when combined with elevated 14-3-3, MRI changes, and clinical symptoms based on CJD diagnostic criteria suggested by Centers for Disease Control and Prevention (CDC) as probable category with myoclonus, extrapyramidal signs, a positive 14-3-3 CSF assay, high signal in caudate/putamen on magnetic resonance imaging (MRI) brain scan or at least two cortical regions (temporal, parietal, occipital) either on diffusion-weighted imaging (DWI) or fluid attenuated inversion recovery (FLAIR) and with ruling out alternative diagnosis which includes vascular causes including ischemic stroke, cerebral venous thrombosis, vasculitis, reversible posterior leukoencephalopathy, autoimmune encephalitis, hypoxic/ anoxic brain injury, osmotic demyelination, metabolic encephalopathy, and mitochondrial diseases.
Conclusion
Our case highlights the clinical dilemma in diagnosing Creutzfeldt–Jakob disease (CJD), as the patient was initially misdiagnosed with status epilepticus due to an unusual presentation. The case demonstrated atypical clinical examination findings and imaging features not typically associated with CJD. This case is particularly distinctive due to the presence of pre-existing epilepsy and prior temporal lobe surgery, which significantly increased the risk of diagnostic anchoring on seizure-related pathology. Several reports describe sCJD presenting as refractory seizures or status epilepticus, often delaying recognition of an underlying prion disease [43-45]. This case underscores the importance of reconsidering the diagnosis when neurological decline persists despite adequate seizure control. This highlights the need for a high level of clinical vigilance for clinicians when managing patients with status epilepticus who fail to respond to conventional antiepileptic therapy, as an alternative diagnosis should be considered.
Funding: No funding or grant has been received for this article.
Conflict of Interest: The authors have no conflict of interest
References
- Uttley, L., Carroll, C., Wong, R., Hilton, D. A., & Stevenson,M. (2020). Creutzfeldt-Jakob disease: a systematic review of global incidence, prevalence, infectivity, and incubation. The Lancet Infectious Diseases, 20(1), e2-e10.
- "Creutzfeldt–Jakob Disease, Classic (CJD) | Prion Diseases| CDC". www.cdc.gov. 1 February 2019. Retrieved 17 June2019. Classic CJD is a human prion disease
- "Creutzfeldt–Jakob Disease Fact Sheet | National Institute of Neurological Disorders and Stroke". NINDS. March 2003. Archived from the original on 4 July 2017. Retrieved 16 July 2017
- Creutzfeldt–Jakob disease @Who Named It
- Department of Health and Human Services, Centers for Disease Control and Prevention, CJD (Creutzfeldt-Jakob Disease, Classic), author 2010. [June 1st 2012].
- Creutzfeldt–Jakob Disease Fact Sheet | National Institute of Neurological Disorders and Stroke. NINDS. March 2003. Archived from the original on 4 July 2017. Retrieved 16 July 2017.
- Creutzfeldt-Jakob Disease Foundation.
- Johnson RT, Gonzalez RG, Frosch MP. Case records of the Massachusetts General Hospital. Case 27-2005. An 80-year-old man with fatigue, unsteady gait, and confusion. N Engl J Med. 2005 Sep 08;353(10):1042-50.
- Ladogana A, Puopolo M, Croes EA, Budka H, Jarius C, Collins S, Klug GM, Sutcliffe T, Giulivi A, Alperovitch A, Delasnerie-Laupretre N, Brandel JP, Poser S, Kretzschmar H, Rietveld I, Mitrova E, Cuesta Jde P, Martinez-Martin P, Glatzel M, Aguzzi A, Knight R, Ward H, Pocchiari M, van Duijn CM, Will RG, Zerr I. Mortality from Creutzfeldt-Jakob disease and related disorders in Europe, Australia, and Canada. Neurology. 2005 May 10;64(9):1586-91.
- Hall WA, Masood W. Creutzfeldt-Jakob Disease. [Updated2025 Jun 23]. In: StatPearls
- Zhu X, Li R, Zhu Y, Tan Y. Diagnostic Challenges in Sporadic Creutzfeldt-Jakob Disease: A Case Study of Typical Clinical Presentation with Negative Findings. Am J Case Rep. 2025 Feb 10;26:e945795..
- "Creutzfeldt–Jakob Disease, Classic (CJD) | Prion Diseases| CDC"
- Hall WA, Masood W. Creutzfeldt-Jakob Disease. [Updated 2025 Jun 23]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-.
- Thompson, A., MacKay, A., Rudge, P., Lukic, A., Porter, M. C., Lowe, J., ... & Mead, S. (2014). Behavioral and psychiatric symptoms in prion disease. American Journal of Psychiatry, 171(3), 265-274.
- Lewis AM, Yu M, DeArmond SJ, et al. Human growth hormone-related iatrogenic Creutzfeldt-Jakob disease with abnormal imaging. Arch Neurol 2006; 63:288.
- Noguchi-Shinohara M, Hamaguchi T, Kitamoto T, et al. Clinical features and diagnosis of dura mater graft associated with Creutzfeldt-Jakob disease. Neurology 2007; 69:360.
- Will, R. G. (2003). Acquired prion disease: iatrogenic CJD,variant CJD, kuru. British medical bulletin, 66(1), 255-265.
- Macfarlane RG, Wroe SJ, Collinge J, et al. Neuroimaging findings in human prion disease. J Neurol Neurosurg Psychiatry 2007; 78:664.
- Chitravas, N., Jung, R. S., Kofskey, D. M., Blevins, J. E., Gambetti, P., Leigh, R. J., & Cohen, M. L. (2011). Treatable neurological disorders misdiagnosed as Creutzfeldt-Jakob disease. Annals of neurology, 70(3), 437-444.
- Rudge, P., Hyare, H., Green, A., Collinge, J., & Mead, S. (2018). Imaging and CSF analyses effectively distinguish CJD from its mimics. Journal of Neurology, Neurosurgery & Psychiatry, 89(5), 461-466.
- Manners, D. N., Parchi, P., Tonon, C., Capellari, S., Strammiello, R., Testa, C., ... & Barbiroli, B. (2009). Pathologic correlates of diffusion MRI changes in Creutzfeldt-Jakob disease. Neurology, 72(16), 1425-1431.
- Fragoso D, Gonçalves Filho A, Pacheco F et al. Imaging of Creutzfeldt-Jakob Disease: Imaging Patterns and Their Differential Diagnosis. Radiographics. 2017;37(1):234-57.
- Cohen OS, Hoffmann C, Lee H, Chapman J, Fulbright RK, Prohovnik I. MRI detection of the cerebellar syndrome in Creutzfeldt-Jakob disease. (2009) Cerebellum (London, England). 8 (3): 373-81.
- Vitali P, Maccagnano E, Caverzasi E et al. Diffusion-Weighted MRI Hyperintensity Patterns Differentiate CJD from Other Rapid Dementias. Neurology. 2011;76(20):1711-9.
- Tschampa HJ, Kallenberg K, Urbach H et-al. MRI in the diagnosis of sporadic Creutzfeldt-Jakob disease: a study on inter-observer agreement. Brain. 2005;128 (9): 2026-33.
- Mauricio Castillo. Neuroradiology Companion. (2011) ISBN:9781451111750
- Steinhoff, B. J., Racker, S., Herrendorf, G., Poser, S., Grosche, S., Zerr, I., ... & Weber, T. (1996). Accuracy and reliability of periodic sharp wave complexes in Creutzfeldt-Jakob disease. Archives of neurology, 53(2), 162-166.
- EEG Presentation in Rapidly Evolving Sporadic Creutzfeldt-Jakob Disease
- Green AJE, Zanusso G. Prion protein amplification techniques. Handb Clin Neurol 2018; 153:357.
- Collie, D. A., Sellar, R. J., Zeidler, M., Colchester, A. C., Knight, R., & Will, R. G. (2001). MRI of Creutzfeldt–Jakob disease: imaging features and recommended MRI protocol. Clinical radiology, 56(9), 726-739.
- Schröter A, Zerr I, Henkel K, et al. Magnetic resonance imaging in the clinical diagnosis of Creutzfeldt-Jakob disease. Arch Neurol 2000; 57:1751.
- Hermann P, Laux M, Glatzel M, et al. Validation and utilization of amended diagnostic criteria in Creutzfeldt-Jakob disease surveillance. Neurology. 2018;91(4):e331-e338.
- Satoh, J. I., Kurohara, K., Yukitake, M., & Kuroda, Y. (1999). The 14-3-3 protein detectable in the cerebrospinal fluid of patients with prion-unrelated neurological diseases is expressed constitutively in neurons and glial cells in culture. European neurology, 41(4), 216-225.
- Muayqil T, Gronseth G, Camicioli R. Evidence-based guideline: diagnostic accuracy of CSF 14-3-3 protein in sporadic Creutzfeldt-Jakob disease: report of the guideline development subcommittee of the American Academy of Neurology. Neurology 2012; 79:1499
- Evidence-based guideline: Diagnostic accuracy of CSF 14-3-3 protein in sporadic Creutzfeldt-Jakob disease: Report of the Guideline Development Subcommittee of the American Academy of Neurology.
- Hsich, G., Kenney, K., Gibbs Jr, C. J., Lee, K. H., & Harrington,M. G. (1996). The 14-3-3 brain protein in cerebrospinal fluid as a marker for transmissible spongiform encephalopathies. New England Journal of Medicine, 335(13), 924-930.
- Matsui, Y., Satoh, K., Miyazaki, T., Shirabe, S., Atarashi, R., Mutsukura, K., ... & Nishida, N. (2011). High sensitivity of an ELISA kit for detection of the gamma-isoform of 14-3-3 proteins: usefulness in laboratory diagnosis of human prion disease. BMC neurology, 11(1), 120.
- Leitão, M. J., Baldeiras, I., Almeida, M. R., Ribeiro, M. H., Santos, A. C., Ribeiro, M., ... & Oliveira, C. R. (2016). Sporadic Creutzfeldt–Jakob disease diagnostic accuracy is improved by a new CSF ELISA 14-3-3γ assay. Neuroscience, 322, 398-407.
- Forner SA, Takada LT, Bettcher BM, et al. Comparing CSF biomarkers and brain MRI in the diagnosis of sporadic Creutzfeldt-Jakob disease. Neurol Clin Pract 2015; 5:116.
- Skillbäck T, Rosén C, Asztely F, et al. Diagnostic performance of cerebrospinal fluid total tau and phosphorylated tau in Creutzfeldt-Jakob disease: results from the Swedish Mortality Registry. JAMA Neurol 2014; 71:476.
- Szabo K, et al. Diffusion abnormalities in seizure disorders. Neurology. 2005;65(11):1696-700.
- Tschampa HJ, et al. Serial MRI in CJD. AJNR Am JNeuroradiol. 2007;28(9):1677-82.
- Kim MO, et al. CJD presenting as refractory status epilepticus.Neurology. 2011;76(10):878-81.
- Monti G, et al. Seizure-onset sporadic CJD. J Neurol.2017;264(4):758-66.
- Chitravas N, et al. Treatable mimics of CJD. Neurology.2011;77(2):111-8