
Current Research in Vaccines Vaccination(CRVV)
ISSN: 2834-880X | DOI: 10.33140/CRVV


Case Report - (2024) Volume 2, Issue 4
Acute Interstitial Nephritis as the First Presenting Feature of Wilson's Disease: A case Report of Diagnostic Dilemma
2Department of Medicine, Mymensingh Medical College, Bangladesh
Received Date: Dec 05, 2023 / Accepted Date: Dec 30, 2023 / Published Date: Dec 30, 2023
Copyright: ©©2024 Ashmita Yadav, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Citation: Yadav, A., Nepali, R. B., Jahangir Alam, A. M. (2024). Acute Interstitial Nephritis as the First Presenting Feature of Wilson's Disease: A case Report of Diagnostic Dilemma. Curr Res Vaccines Vaccination, 2(4), 95-98.
Abstract
Introduction: Wilson's disease( also called as hepaticolenticular degeneration) is a rare genetic (autosomal recessive) disorder resulting from mutation in ATP7B gene presenting with features affecting various organs of the body namely( liver, brain, kidney, cornea) due to deposition of excess copper in these organs. It can lead to fatal consequences if not diagnosed and treated in time however prompt diagnosis and treatment can prevent further deterioration of symptoms.
Case Presentation: The authors hereby present a case of 11 years old male who presented with symptoms mimicking post streptococcal glomerulonephritis (PSGN) however after a thorough history , clinical examination findings and detailed investigations he was diagnosed as a case of decompensated chronic liver disease (CLD) due to Wilson’s disease with acute interstitial nephritis(AIN).
Discussion: He has been on chelating agent, zinc acetate and has been asked to avoid copper containing foods. The patient has been followed up regularly since the beginning of treatment.
Conclusion: The main objective of the authors is to highlight a rare case of wilson's disease whose first presentation was renal involvement which led to diagnostic dilemma; further evaluation and investigations ultimately established the diagnosis.
Keywords
Wilson’s, AIN, Manifestations, Copper, Chelating Agents
Introduction
Wilson's disease(WD) also known as hepaticolenticular degeneration is rare genetic disorder((autosomal recessive) caused by a mutation in ATP7B gene that affects 1 in 30,000 population. It is characterised by inability of liver to eliminate copper from the body and build-up of extra copper in the organs such as liver, brain, kidney and cornea [1,2]. It can affect any age group the most common being 5-35 years of age and the incidence is equal in males and females [2]. Signs and symptoms of WD are variable depending on the organs/parts of body affected; Presentations include -Yellowish discoloration of sclera, skin or whole body, swelling and pain in the body ,fatigue, nausea ,loss of appetite and bleeding tendency. Psychiatric manifestations include depression , anxiety , mood swings ,disruption of thoughts and feelings (psychosis),tremors, rigidity ,difficulty speaking and swallowing , problems with balance and walking, seizure and personality changes [3]. Renal manifestations ranges from acute interstitial nephritis, oliguria, renal tubular acidosis and nephrolithiasis to fanconi's anaemia [4,5]. The excessive copper first accumulates in liver and gradually seeps into other organs and tissues like subthalamus, putamen, cortex of brain, kidneys and cornea [4]. There is no completely reliable investigation for the diagnosis of WD however low ceruloplasmin and high 24hrs urinary copper with KF(Kayser-Fleischer) rings are sufficient for diagnosis [6]. KF rings are dark rings encircling the cornea of the eye due to copper deposition in Descemet's membrane as a result of WD [7]. Liver biopsy is considered as the gold standard for diagnosis and should be carried out if alternate diagnosis are considered.
MRI of brain is a very valuable and relevant investigation when a patient presents with neurological manifestations which mostly affects putamen, pons, midbrain and thalamus. The diagnosis can also be supported by mutation analysis is ATP7B gene [8]. Treatment options include chelating agents such as
a) D penicillamine : It is a copper chelating agent which also stops pyridoxine from working requiring pyridoxine supplemention .
b) Trientene : It has a similar working mechanism as penicillamine but can worsen neurological features .
c) Zinc acetate: It prevents absorption of copper from food and
d) Avoidance of food containing copper [6].
Untreated patients have a poor prognosis with low life expectancy however prompt diagnosis and treatment can help patients achieve a normal life expectancy [9] .
Case Report
We hereby report a case of 11 years old male who presented to us with reddish discoloration of urine for 1 day and scanty micturition . He also mentioned of fever 2 weeks back which was high grade not associated with chills and rigor relieved by medication , subsided with sweating but the highest temperature wasn't recorded. He denied any cough, sore throat or skin infection/ lesions. He also complained of swelling of the body for 1 month which first started in the abdomen and gradually progressed to involve the face and the legs, not associated with any abdominal pain but with abdominal discomfort and heaviness.
He also noticed gradual yellowish discoloration of sclera and skin of whole body. He mentioned one previous episode of jaundice 2 months back. There was no history of hematemesis, melena ,loss of consciousness, nausea, vomiting ,loss of appetite or shortness of breath.
There was no behavioural disturbance , mood or personality changes ,seizure or any feature of psychosis. The parents denied any history of repeated blood transfusion, use of herbal medication or IV drug abuse/sharing of needles.
There was no history of consanguineous marriage of his parents and his other sibling is in good health.
On Examination
He was ill looking, mildly anaemic and icteric, pitting pedal edema was evident , no other stigmata of CLD could be found.
Examination of the gastrointestinal system revealed presence of shifting dullness suggestive of ascites. Spleen was enlarged and was 4cm from the left costal margin along the anterior axillary line towards the right iliac fossa , surface was smooth, firm in consistency and non-tender with palpable splenic notch and no rub could be heard. No other organs were palpable.
Neurological status of the patient was normal and examination revealed no abnormality. Slit lamp examination revealed bilateral KF rings.
|
PARAMETER |
VALUES |
|
Hb% |
8.7 g/dl |
|
RBC |
3.10×10^6/uL |
|
WBC |
6.06×10^3/uL |
|
Platelet |
97,000/mm^3 |
|
S.creatinine |
0.7 mg/dl |
|
s.bilirubin |
3.9 mg/dl |
|
SGPT(ALT) |
73 U/L |
|
S.Albumin |
3.5g/ dl |
|
Ceruloplasmin |
60 mg/dl (ref-200-600) |
|
24hrs urinary copper |
254 microgram/day(ref- 30 -50) |
|
INR |
2.19 |
|
Prothrombin time |
26 seconds |
|
URINE R/M/E Pus cells RBC |
4-5/HPF 1 -2 /HPF |
Table 1: Investigation and Diagnosis
USG of Whole Abdomen Cirrhosis of the liver with mild splenomegaly and very mild ascites. Pelvic dilatation in left kidney Sludge ball in gall bladder. Endoscopy of UGIT: Normal findings

Figure 1: Endoscopy of Upper Gastrointestinal System
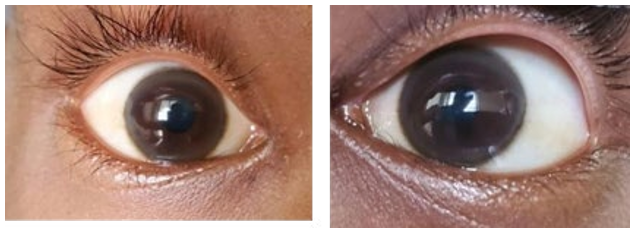
Figure 2: KF Ring (Right Eye) Figure 3: KF Ring(Left Eye)
He was eventually diagnosed as a case of Decompensated CLD due to Wilson’s disease with AIN.
Discussion
Wilson's disease is a rare genetic disorder and can cause fatal consequences if not diagnosed and treated in time .Our patient presented with features mimicking PSGN giving a history of high colored urine and oliguria which was preceded by a episode of fever 2 weeks back. There was no behavioural disturbance, tremor , mood or personality changes. However 2 episodes of jaundice and swelling of the body which first started in the abdomen gradually progressing to involve face and limbs led to the suspicion of CLD. We run the liver function tests and found they were severely deranged. Ultrasonogram of the abdomen revealed cirrhosis with mild splenomegaly and mild ascites. We performed endoscopy to see any variceal bleeding which demonstrated normal findings. C3 level was done to rule out PSGN and it was normal in range. Slit lamp examination revealed bilateral KF rings and though there were no neurological manifestations we screened him for WD which revealed serum ceruloplasmin was low and 24hrs urinary copper was high. This investigation result alongside bilateral KF rings helped us reach our diagnosis.
The high colored urine and oliguria were suggestive of AIN as a consequence of WD .The patient was started on D penicillamine 250mg BD and pyridoxine supplemention was done along with it. He was also given lactulose as a preventive measure for development of features of encephalopathy.
Zinc acetate has been prescribed and he has been advised to avoid copper containing foods i.e. shellfish, liver, chocolate, dried fruits, dried peas and beans, mushroom and nuts. The other apparently well sibling has also been asked to get screened for WD.
Conclusion
A rare yet fatal disease entity affecting 1 in 30,000 people of any age group presenting with multiorgan involvement can be diagnosed clinically and with the help of relevant investigations. The diagnosis can also be supported by mutation analysis if facilities for the same are available. WD should be suspected in a patient presenting with any of the symptoms and signs mentioned above. Renal manifestations though not very common but can range from AIN, oliguria, nephrolithiasis, renal tubular acidosis to fanconi's anaemia. Prompt therapy with copper chelating agents alongside dietary restrictions of copper can help prevent further deterioration of the disease. The dosages of medications can be adjusted on subsequent follow ups and being an autosomal recessive disorder the siblings should also be encouraged to get screened for the disease.
Conflict of Interest
None
Funding
None
References
- Krause L. (2018). Wilson’s disease: Risk factors, causes, & symptoms.
- Wilson’s disease (2023). Mayo Clinic.
- Wilson’s Disease symptoms & treatment. Children’s Hospital of Pittsburgh.
- Chaudhry, H. S., Anilkumar, A. C. (2023). Wilson Disease. StatPearls Publishing.
- Schilsky, M. L., & Mistry, P. K. (2009). Wilson Disease and the Kidney. In Genetic Diseases of the Kidney (pp. 709-713). Academic Press.
- Wilson’s disease (2023). Mayoclinic.org.
- Wikipedia contributors. Kayser–Fleischer ring.
- Singh P, Ahluwalia A, Saggar K, Grewal CS. (2011). Wilson's disease: MRI features. J Pediatr Neurosci, 6(1):27-8.
- Naqvi E. (2022). Wilson disease prognosis. Rare Disease Advisor.