
Annals of Computational Physics and Material Science(ACPMS)
ISSN: 2997-2795 | DOI: 10.33140/ACPMS


Research Article - (2025) Volume 2, Issue 1
The Completed Density Functional Theory (cDFT) for Accurate Description and Prediction of Properties of Materials
2Department of Studies and Research (DSR) in Physics, Center of Calculation, Modeling and Simulation , Mali
3Department of Physics, Materials – Composite Systems and Applications (MASCA), Cheikh Anta Diop Univ, Senegal
Received Date: Dec 21, 2024 / Accepted Date: Jan 08, 2025 / Published Date: Jan 13, 2025
Copyright: ©Â©2025 Diola Bagayoko, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Citation: Bagayoko, D., Diakite, Y. I., Dioum, A., Malozovsky, Y. (2025). The Completed Density Functional Theory (cDFT) for Accurate Description and Prediction of Properties of Materials. Ann Comp Phy Material Sci, 2(1), 01-05.
Abstract
The foundational theorems of DFT require, for its correct application, the use of the ground state charge density of a material for calculating its electronic and related properties. The à priori unknown nature of this ground state charge density points to the incomplete nature of the seminal DFT. Mainstream calculations have mostly assumed that results obtained with self-consistent iterations using a single basis set represent the ground state of a material; such results are stationary states among an infinite number of such states – with no relation to the ground state of the material under study. The Completion of DFT [AIP Advance, 4, 127104 (2014)] entailed (a) the introduction of the second corollary to the first DFT theorem and (b) the methodical search for and attainment of the ground state of a material with successive, self-consistent calculations with progressively augmented basis sets. With (a) and (b), the completed density functional theory (cDFT) has unfailingly and accurately predicted properties of several materials and described electronic and related properties of dozens of semiconductors, including their band gaps. The cDFT does not invoke a self-interaction correction or a derivative discontinuity of the exchange-correlation energy. It does not utilize ad hoc potentials. Results of cDFT calculations possess the full physical content of the theory and are in accord with corresponding, experimental ones; they clearly indicate that objectives of the Materials Genome Initiative (MGI) can be reached with a widespread utilization of cDFT [MRS Advances 8, 619-625 (2023)].
Keywords
Completed Density Functional Theory (cDFT), Band Gaps, Ground State, Generalized Minimization of the Energy Functional
Introduction
The first foundational theorem of density functional theory, as stated by Rothenberg and Kohn, follows [1]. “Let an arbitrary number of electrons in a box be subject to an external potential and the electron-electron Coulomb repulsion, then, this potential is a unique functional of the electron density, except for an additive constant.” The first corollary of this theorem is that the energy functional, Ev[Ψ’] = (Ψ’,HΨ’), is a unique functional of the electron density. This energy is the sum of the occupied energies in the spectrum of the Hamiltonian. In 2014, Bagayoko introduced the second corollary of the above theorem, namely, that the spectrum of the Hamiltonian is a unique functional of the electron charge density [2]. The second DFT theorem, also known as the DFT variational principle, states that the energy functional reaches its minimum when the electron density is that of the ground state. Specifically, according to Hohenberg and Kohn, “It is well known that for a system of N particles, the energy functional of Ψ′
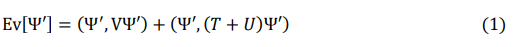
has a minimum at the correct ground state Ψ, relative to arbitrary variations of Ψ′ in which the total number of particles is kept constant.” In the formula, the theorem is:
∫ v(r)n'(r)dr + F[n'] > ∫ v(r)n(r)dr + F[n] (2)
where n(r) is the ground state density and n’(r) is any other density different from n(r). Let us note that the same potential v(r) is on both sides of the inequality. Any reasonable basis set Ψ’, upon reaching self-consistency, leads to a corresponding electron density n’(r). “Reasonable” in this context means that the referenced basis set accounts for all the electrons in the system. For any given system, there exists an infinite number of reasonable basis sets. Each one of them produces a corresponding charge density upon the attainment of self-consistency:

density functional theory for predicting or describing electronic and related properties of materials has to utilize the ground state charge density in computations. Unfortunately, the 3-dimensional ground state charge densities of atoms, molecules, semiconductors, insulators, and metals are à priori unknown. Throughout the world, researchers have been following a suggestion made by Kohn and Sham [3]. It consists of judiciously selecting a basis set to perform self-consistent iteration in order to produce results that are taken to represent those of the ground state. Unfortunately, a state of a material obtained with self-consistency iterations with a single basis set is a stationary one among an infinite number of such states. It should be noted that before the above suggestion, Kohn and Sham emphatically stated that the four equations defining the local density approximation (LDA) are the ones to be solved simultaneously [3]. Once the expression of the exchange- correlation energy is known, its functional derivative provides the potential. Then, the four equations are reduced to the Kohn- Sham equation and the one giving the charge density in terms of the wave functions of the occupied states, as in Equation 3. With the knowledge of the Rayleigh theorem for eigenvalues, solving these two equations simultaneously requires the variation of the size of the basis set, along with its radial features and angular richness [4,5]. The “arbitrary variations” in the second theorem are constrained only by the “reasonableness” defined above. Almost universally, results of calculations using a single basis set have disagreed with experiments for atoms, molecules, semiconductors, and insulators –for the energy or band gaps. Specifically, they generally underestimate by 30-50% or more the measured energy gaps (for finite systems) and band gaps for crystalline semiconductors and insulators.
The Completed DFT (cDFT)
In 1998, Bagayoko and collaborators [4] introduced a computational method radically different from the mainstream utilization of a single basis set for DFT calculations. In 1999, Zhao, Bagayoko, and Williams applied the method to study successfully GaN, carbon, silicon, and RuO2 [5]. The resulting Bagayoko, Zhao, and Williams (BZW) method was inspired by Bagayoko’s utilization of the Rayleigh theorem for eigenvalues in performing calculations with contracted basis sets [6]. This theorem states that when the same eigenvalue equation is solved with N and (N+1) calculations with augmented basis sets up to the point where three
(3) consecutive ones produce the same occupied energies. Those stable occupied energies represent the true ground state of the system under study. At first, the Bagayoko, Zhao, and Williams (BZW) method augmented a basis set by adding orbitals in the increasing order of the excited energies they represent. Subsequent works by Ekuma and Franklin added orbitals differently: For a given quantum number n, the orbitals are added in the p, d, f, and s order, if applicable, instead of that of increasing energy [7,8]. “If applicable” means that said orbital was occupied in one of the neutral atoms in the system under study. This enhancement by Ekuma and Franklin led to the BZW-EF method. This counter- intuitive approach led to descriptions and predictions in excellent agreement with experimental findings. This “excellent agreement” stems in part from the fact that for valence electrons, each one of which is under the influence two or more atomic or ionic sites, polarization has primacy over spherical symmetry. Further examination of the original DFT paper revealed that, indeed, one could have “arbitrary variations” of the reasonable basis set provided the total number of particles is kept constant - according to the second DFT theorem [1].
The above process of the BZW and BZW-EF method completes DFT by providing a rigorous algorithm for finding the true ground state of the system under study. The first basis set to produce the ground state, as confirmed by two subsequent calculations, is the one providing the completed DFT (cDFT) description of the material under study. The basis set for this calculation is known as the optimal basis set. Upon the attainment of self-consistency, it produces the true ground state charge density. The calculations following it also yield the ground state. However, any eigenvalue produced by these subsequent calculations and that is not obtained with the optimal basis set, does not belong to the spectrum of the Hamiltonian, a unique functional of the ground state charge density - as per the second corollary of the first DFT theorem.
Recommendations For Using cDFT
There are distinct issues that mainstream condensed matter theory community should address to realize the presently missed benefits of cDFT, including the attainment of the objectives of the Materials Genome Initiative (MGI) [9].
cDFT Functional
The first of these issues is the recognition that while the exact functional of DFT (or cDFT) is not known, good to excellent approximations exist that are not presently utilized properly.
Indeed, even the exact functional will lead to energy gaps (for finite systems) or band gaps (for crystalline systems) that disagree with corresponding experimental findings - as long as the calculations do not verifiably reach the ground states of materials under study. Specifically, the Ceperley and Alder functional, with the parameterization of Vosko, Wilk, and Nusair (VWN), has led to accurate descriptions of electronic and related properties of more than 30 semiconductors [10,11]. With the VWN potential, cDFT has correctly described these materials and predicted band gaps and other properties for several semiconductors [2]. In the spectacular case of rutile TiO2, our prediction of an indirect band gap, subsequently confirmed, was contrary not only to findings from mainstream DFT calculations but also to experimental results [12].
Ad hoc Potentials
The condensed matter theory community, for the second issue, could dramatically reduce the use and the development of ad hoc potentials. While these potentials have served us well in describing some materials, it should be recognized that they have no predictive capacity. Indeed, as underscored by Bagayoko, any potential not straightforwardly obtained as the functional derivative of an exchange-correlation energy is not entirely a DFT potential [2]. We posit that the need for ad hoc potential stemmed from the failure of calculations using the incomplete DFT, i.e., ones where iterations with a single basis set are erroneously presumed to lead to the ground states of materials under study. In light of the highly accurate results from cDFT, as illustrated elsewhere and below, the current investment of talents, efforts, and funds in the development of ad hoc potential should be drastically reduced [2,13]. Medvedev et al., noted that “density functional theory is straying from the path toward an exact functional [14].” Indeed, after examining some 128 functionals, they found that up to the year 2000, densities resulting from developed functionals were closer to corresponding, exact ones. After 2000, however, “this trend was reversed by unconstrained functionals sacrificing physical rigor for the flexibility of empirical fitting [14].”
The Need for Exponential and Gaussian Functions and the Attainment of the Ground State
The angular richness rendered by spherical or cubic harmonics cannot be obtained with plane waves. Additionally, the adequacy of the radial parts of exponential or Gaussian orbitals in describing materials cannot be equaled by plane waves. Consequently, the full implementation of cDFT requires software packages utilizing
(a) full potentials (b) exponential or Gaussian orbitals, and (c) the flexibility of augmenting the basis set, one orbital at a time, to reach the true ground state of a material under study. While pseudopotential and plane wave program packages have served the community well, obtaining calculated results that possess the full physical content of cDFT and agree with corresponding, experimental ones requires (a), (b), and (c) as spelled out above. Several of our cDFT calculations provide a table showing the successive augmentation of the basis set, one orbital at a time, for the consecutive, self-consistent calculations [7,8]. The highest number of such calculations for all the materials we have studied to date is eight (8). For several semiconductors, this number is between five (5) and seven (7). While mainstream calculations utilize iterations with a single basis set to reach stationary states, the noted augmentation of the basis set lowers the occupied energies down to the ground state of a material. With machine learning (ML), the referenced augmentation of the basis set can be significantly facilitated, even for large molecules with different atomic species or supercell calculations for materials with no translation symmetry.
The Role of Funding Sources and of Journals
Most reputable funding sources and journals have knowledgeable program officers and manuscript referees, respectively. If public and private funding sources do not encourage the development of software packages that permit cDFT calculations, the attainment of the objectives of the Materials Genome Initiative (MGI) may remain in doubt. Similarly, the funding of calculations that adhere to (a) through (c) above will be necessary for significant progress in the realization of the potentials of cDFT in accurately describing and predicting properties of materials, including organic ones.
In 2016, Bagayoko published the correct understanding and the completion of the relativistic generalization of density functional theory by Rajagopal and Callaway [15,16]. As was the case for non-relativistic DFT, the correct application of the relativistic one requires the verifiable attainment of the ground state – with successive augmentation of the four (4) component spinors basis set. Upon reaching the true ground state, self-consistent iterations lead to the ground state current density whose fourth component gives the ground state charge density. Presently, we do not know of a single semi-relativistic program package, let alone a fully relativistic one, that permits the successive augmentation of the spinor basis set to search for the true ground state. The development of a fully relativistic program package for electronic structure calculations, either for atoms, molecules, or for solids, is expected to require a significant funding. Much remains to be theoretically understood about the electronic and related properties of heavy elements and of materials containing them, including several high-temperature superconductors.
Journal editors and manuscript referees may have to abandon some beliefs that arose as the result of the incorrect application of the incomplete, original density functional theory. Indeed, the derivative discontinuity of the exchange-correlation energy was partly introduced to remedy the failure of mainstream calculations to produce band gaps that agree with corresponding, experimental ones. Many of these mainstream band gaps are 30 to 50% or more smaller than the corresponding measured ones. As noted above, cDFT calculations, by strictly adhering to the fundamental theorems of DFT, produce the actual ground states of the systems under study. In doing so, they obtain results that possess the full physical content of DFT and agree with corresponding experimental ones.
Illustrative Results from cDFT
With the BZW and BZW-EF methods, we have correctly predicted electronic and related properties of cubic silicon nitride rutile TiO2 [12], (c-Si3N4), cubic indium nitride (c-InN), wurtzite InN and cubic magnesium silicide (Mg2Si) [17-20]. Of course, the band gap is a central electronic property these calculations accurately predicted. The table below provides illustrative examples of results for small to large band gaps semiconductors and BeO, an insulator.
|
Material |
DFT Potential |
Number of Calculated Gaps |
Ranges of Calculated Gaps |
Experimental Gaps |
DFT BZW or BZW-EF Gaps |
|
Cubic InN |
LDA & GGA |
10 |
-0.55 - + 0.08 |
0.61 eV |
0.65 eV18 |
|
c-Mg2Si |
LDA &GGA |
10 |
0.12 – 0.42 |
0.65 – 0.80 eV |
0.89 eV20 |
|
w-AlN |
LDA & GGA |
11 |
3.9 – 4.78 eV |
6.2 – 6.2 ±0.2 eV |
6.28 eV21 |
|
zb-ZnS |
LDA & GGA |
5 |
1.65 – 2.37 eV |
3.723 eV |
3.725 eV22 |
|
w-GaN |
LDA & GGA |
17 |
1.68 – 2.52 eV |
3.3-3.5 eV |
3.20 & 3.29 eV23 |
|
w-BeO |
LDA |
9 |
7.0 – 7.8 eV |
10.24 – 10.63±0.10 eV |
10.3 eV24 |
|
Rutile TiO2 |
LDA & GGA |
18 |
1.67 – 2.12 eV |
3.00 – 3.10 eV |
2.95 & 3.05 eV12 |
|
w-ZnO |
LDA |
12 |
0.23 – 2.26 eV |
3.30 – 3.40 eV |
3.39 eV8 |
|
zb-BP |
LDA & GGA |
15 |
1.11 – 1.38 eV |
2.02±0.05 eV |
2.02 eV25 |
|
c-BN |
LDA & GGA |
9 |
4.20 – 4.47 eV |
6.20 – 6.4±0.5 eV |
6.48 eV26 |
Table: Band gaps from ab initio, self-consistent DFT calculations in the literature for the identified materials. The number of calculations before our cDFT ones for each material is in Colum 3. Column 4 shows the range of the band gap values obtained by these previous DFT calculations. These previous values are all underestimates of the corresponding experimental values in Column 5, which agree with our cDFT results in Column 6
Once a calculation leads to an erroneous band gap, it can no longer continue to obtain accurate dielectric or optical properties. The effective mass for the electron is highly sensitive to the curvature around the minimum of the unoccupied bands. With the correct electronic structure and related wave functions, most other properties of materials can be studied theoretically. Several publications from our group, as per their titles, provide predictions of electronic and related properties of materials. We urge readers to consult AIP Advances 2014, MRS Advances 2023, and the 2024 Proceedings of MSAS for numerous examples of accurate predictions or descriptions of electronic and related properties of materials, with emphasis on semiconductors [2,13,21].
Conclusion
We note that the above results, and the many others in the indicated publications, were obtained using cDFT. With successively augmented basis sets, we searched for and verifiably reached the ground states of the materials under study. In doing so, we did not need to invoke self-interaction correction or the derivative discontinuity of the exchange-correlation energy. As for the latter, Bagayoko proved that it cannot be relevant to a solution of the Kohn-Sham equation [2]. Indeed, the derivative discontinuity was derived for semiconductors and for insulators using systems whose total numbers of particles were not constant; the Kohn- Sham equation is obtained by setting the variation of the total number of particles equal to zero! Bagayoko added that while the referenced derivative discontinuities may be useful in some ways, they have no bearing on the calculated band gaps of materials. The quasi-universal underestimation of energy and band gaps by mainstream calculations stems squarely from their utilization of a single basis set to perform self-consistent calculations whose stationary states are mistakenly taken to be the ground state of the materials [22-27].
Acknowledgement
This work was funded in part by the US Department of Energy, National Nuclear Security Administration (NNSA) (Awards No. DE- NA0003679 and No. DE-NA0002630); the National Science Foundation (NSF) (Awards No. EPS- 1003897 and No. HRD-1503226); the Department of the Navy, Office of Naval Research (ONR), through the Timbuktu Academy, (www.subr. edu/TimbuktuAcademy) (Grants No. N00014-93-1-1368 and No. N00014-98-1-0748); NASA, through LaSPACE (Award No. 5-27316), LONI; SUBR; the Ministry for Higher Education and Scientific Research of Mali; and Cheikh Anta Diop University (UCAD), Dakar, Senegal.
References
1. Hohenberg, P., & Kohn, W. (1964). Inhomogeneous electron gas. Physical review, 136(3B), B864.
2. Bagayoko, D. (2014). Understanding density functional theory (DFT) and completing it in practice. AIP Advances, 4(12).
3. Kohn, W., & Sham, L. J. (1965). Self-consistent equations including exchange and correlation effects. Physical review, 140(4A), A1133.
4. Bagayoko, D., Zhao, G. L., Fan, J. D., & Wang, J. T. (1998). Ab initio calculations of the electronic structure and optical properties of ferroelectric tetragonal. Journal of Physics: Condensed Matter, 10(25), 5645.
5. Zhao, G. L., Bagayoko, D., & Williams, T. D. (1999). Local- density-approximation prediction of electronic properties of GaN, Si, C, and RuO 2. Physical Review B, 60(3), 1563.
6. Bagayoko, D. (1983). Contraction of gaussian basis sets and the total energy of fcc copper. International Journal of Quantum Chemistry, 24(S17), 527-535.
7. Ekuma, C. E., Jarrell, M., Moreno, J., & Bagayoko, D. (2013). Re-examining the electronic structure of germanium: A first- principle study. Physics Letters A, 377(34-36), 2172-2176.
8. Franklin, L., Ekuma, C. E., Zhao, G. L., & Bagayoko, D. (2013). Density functional theory description of electronic properties of wurtzite zinc oxide. Journal of Physics and Chemistry of Solids, 74(5), 729-736.
9. The Materials Genome Initiative.
10. Ceperley, D. M., & Alder, B. J. (1980). Ground state of the electron gas by a stochastic method. Physical review letters, 45(7), 566.
11. Vosko, S. H., Wilk, L., & Nusair, M. (1980). Accurate spin- dependent electron liquid correlation energies for local spin density calculations: a critical analysis. Canadian Journal of physics, 58(8), 1200-1211.
12. Ekuma, C. E., & Bagayoko, D. (2011). Ab-initio electronic and structural properties of rutile titanium dioxide. Japanese Journal of Applied Physics, 50(10R), 101103.
13. Bagayoko, D., & Diakité, Y. I. (2023). Realizing the potentials of density functional theory (DFT) and of the materials genome initiative (MGI). MRS Advances, 8(11), 619-625.
14. Medvedev, M. G., Bushmarinov, I. S., Sun, J., Perdew, J. P., & Lyssenko, K. A. (2017). Density functional theory is straying from the path toward the exact functional. Science, 355(6320), 49-52.
15. Bagayoko, D. (2016). Understanding the relativistic generalization of density functional theory (DFT) and completing it in practice. Journal of Modern Physics, 7(9), 911-919.
16. Rajagopal, A. K., & Callaway, J. (1973). Inhomogeneous electron gas. Physical Review B, 7(5), 1912.
17. Bagayoko, D., & Zhao, G. L. (2001). Predicted electronic properties of cubic Si3N4. Physica C: Superconductivity and its applications, 364, 261-264.
18. Bagayoko, D., Franklin, L., & Zhao, G. L. (2004). Predictions of electronic, structural, and elastic properties of cubic InN. Journal of Applied Physics, 96(8), 4297-4301.
19. Bagayoko, D., & Franklin, L. (2005). Density-functional theory band gap of wurtzite InN. Journal of applied physics,97(12).
20. Dioum, A., Diakité, Y. I., Malozovsky, Y., Ayirizia, B. A., Beye, A. C., & Bagayoko, D. (2023). First-principles investigation of electronic and related properties of cubic magnesium silicide (Mg2Si). Computation, 11(2), 40.
21. Nwigboji, I. H., Ejembi, J. I., Malozovsky, Y., Khamala, B., Franklin, L., Zhao, G., ... & Bagayoko, D. (2015). Ab- initio computations of electronic and transport properties of wurtzite aluminum nitride (w-AlN). Materials Chemistry and Physics, 157, 80-86.
22. Khamala, B., Franklin, L., Malozovsky, Y., Stewart, A., Saleem, H., & Bagayoko, D. (2015). Computational Condensed Matter.
23. Diakité, Y. I., Traoré, S. D., Malozovsky, Y., Khamala, B., Franklin, L., & Bagayoko, D. (2014). Calculated electronic and related properties of wurtzite and zinc blende gallium nitride (GaN). arXiv preprint arXiv:1410.0984.
24. Bamba, C. O., Inakpenu, R., Diakite, Y. I., Franklin, L., Malozovsky, Y., Stewart, A. D., & Bagayoko, D. (2017). Accurate electronic, transport, and related properties of wurtzite beryllium oxide (w-BeO). Journal of Modern Physics, 8(12), 1938.
25. Ejembi, J. I., Nwigboji, I. H., Franklin, L., Malozovsky, Y., Zhao, G. L., & Bagayoko, D. (2014). Ab-initio calculations of electronic, transport, and structural properties of boron phosphide. Journal of Applied Physics, 116(10).
26. Stewart, A., Hart, D., Khamala, B., Malovzowsky, Y., & Bagayoko, D. (2014). Ab-Initio Calculations of the Electronic, Transport, and Bulk Properties of Cubic Boron Nitirde. Journal: JOURNAL OF ADVANCES IN PHYSICS, 9(1), 2277- 2286.
27. Bagayoko, D., Diakité, Y. I., Malozovsky, Y. (2024). Complemented Density functional theory (cDFT) for the accurate description and prediction of material properties, Société Malienne des Sciences Appliquées – Refereed Proceedings of 14th MSAS Conference -Bamako, 292-296.